Nice Mn metallocenes for someone interesting in doing Mn-55 NMR
Chemistry - A European Journal Volume: 12, Issue: 11 April 3, 2006
Structural and Magnetic Studies of the Tris(cyclopentadienyl)manganese(II) “Paddle-Wheel” Anions [Cp3−n(MeCp)nMn]− (n=0–3, MeCp=C5H4CH3, Cp=C5H5) II) “Paddle-Wheel” Anions [Cp3−n(MeCp)nMn]− (n=0–3, MeCp=C5H4CH3, Cp=C5H5)
Alvarez, Carmen Soria; Bashall, Alan; McInnes, Eric J. L.; Layfield, Richard A.; Mole, Richard A.; et. al. pp. 3053 - 3060
Abstract:The ion-contact complexes [{(h 5 -Cp)2 Mn(h 2 :h 5 -Cp)K}3 ]·0.5 THF (1 ·0.5 THF)and [{(h 2 -Cp)2 (h 2 ;h 5 -MeCp)MnK(thf)}]·2 THF (2 ·2 THF) and ion-separated complexes [Mg-(thf)6 ][(h 2 -Cp)3 Mn ]2 (3 ),[Mg(thf)6 ][(h 2 -Cp)(h 2 -MeCp)2 Mn)]2 ·0.5 THF (4 ·0.5 THF),[Mg(thf)6 ][(h 2 -MeCp)3 Mn)]2 ·0.5 THF (5 ·0.5 THF) and[Li([12 ]crown-4)]5 [(h -Cp)3 Mn ]5 (6 )(Cp =C5 H5 ,CpMe =C5 H4 CH3 ),have been prepared and structurally characterised. The effects of varying the Cp and CpMe ligands in complexes 1 –5 have been probed by variable-temperature magnetic susceptibility measurements and EPR spectroscopic studies.
Monday, May 29, 2006
Chemistry - A European Journal Volume: 12, Issue: 11
Tris(pyridylmethylamino)cyclotriguaiacylene Cavitands: An Investigation of the Solution and Solid-State Behaviour of Metallo-Supramolecular Cages and Cavitand-Based Coordination Polymers.
Sumby, Christopher J.; Fisher, Julie; Prior, Timothy J.; Hardie, Michaele J. pp. 2945 - 2959
Abstract:The synthesis of the three isomeric tris(pyridylmethylamino)cy-clotriguaiacylene cavitands is reported, along with the crystal structures of the 2-and 4-pyridyl derivatives.The gen-erality of a previously described [Ag2 {tris(3-pyridylmethylamino)cyclo- triguaiacylene}2 ]2 +dimeric capsule motif and th [Ag4 {tris(4-pyridylm thyl- amino)cyclotriguaiacylene}4 ]4 +tetrahe-dron with several silver salts was confirmed in the solid state and the corre-sponding solution species were investi-gated by NMR spectroscopy.Host –guest interactions in thesesystems have been probed and these interactions are demonstrated to alter and in-fluence the self-assembly outcome of the reaction.Notably,introduction of larger glutaronitrile guest molecules to the [Ag4 L4 ]4 +tetrahedron system pre-vents formation of the tetrahedral
structure,resulting instead in the for-mation of a 4.8 2 coordination network in the solid state.In the absence of templating acetonitrile guests in the [Ag2 (3 )2 ]2 +capsule system,formation of a cage-based one-dimensional coor-dination polymer is observed.
Tris(pyridylmethylamino)cyclotriguaiacylene Cavitands: An Investigation of the Solution and Solid-State Behaviour of Metallo-Supramolecular Cages and Cavitand-Based Coordination Polymers.
Sumby, Christopher J.; Fisher, Julie; Prior, Timothy J.; Hardie, Michaele J. pp. 2945 - 2959
Abstract:The synthesis of the three isomeric tris(pyridylmethylamino)cy-clotriguaiacylene cavitands is reported, along with the crystal structures of the 2-and 4-pyridyl derivatives.The gen-erality of a previously described [Ag2 {tris(3-pyridylmethylamino)cyclo- triguaiacylene}2 ]2 +dimeric capsule motif and th [Ag4 {tris(4-pyridylm thyl- amino)cyclotriguaiacylene}4 ]4 +tetrahe-dron with several silver salts was confirmed in the solid state and the corre-sponding solution species were investi-gated by NMR spectroscopy.Host –guest interactions in thesesystems have been probed and these interactions are demonstrated to alter and in-fluence the self-assembly outcome of the reaction.Notably,introduction of larger glutaronitrile guest molecules to the [Ag4 L4 ]4 +tetrahedron system pre-vents formation of the tetrahedral
structure,resulting instead in the for-mation of a 4.8 2 coordination network in the solid state.In the absence of templating acetonitrile guests in the [Ag2 (3 )2 ]2 +capsule system,formation of a cage-based one-dimensional coor-dination polymer is observed.
Thursday, May 25, 2006
CPL: Ashbrook and Wimperis: 2H DQMAS for monitoring chemical dynamics
Chemical Physics Letters
Volume 423, Issues 4-6 , 1 June 2006, Pages 276-281
2H double-quantum MAS NMR spectroscopy as a probe of dynamics on the microsecond timescale in solidsstar, open
Marica Cutajar, Sharon E. Ashbrook and Stephen Wimperis
a Department of Chemistry and WestCHEM, University of Glasgow, Glasgow G12 8QQ, United Kingdom
b School of Chemistry and EastCHEM, University of St Andrews, St Andrews KY16 9ST, United Kingdom
Abstract
The sidebands observed in the magic angle spinning (MAS) NMR spectrum of a spin I = 1 2H nucleus may be very strongly broadened if motion is present in the solid. The broadening arises from interference between the line-narrowing effects of MAS and the dynamics-driven reorientation of the 2H quadrupole tensor. Here, we show that this motional broadening is absent or much reduced in the corresponding double-quantum MAS NMR spectrum. This observation suggests a new approach to studying dynamics in solids and examples are drawn from 2H MAS NMR of oxalic acid dihydrate, sodium tetrathionate dihydrate, and the synthetic polymer PMMA.
Volume 423, Issues 4-6 , 1 June 2006, Pages 276-281
2H double-quantum MAS NMR spectroscopy as a probe of dynamics on the microsecond timescale in solidsstar, open
Marica Cutajar, Sharon E. Ashbrook and Stephen Wimperis
a Department of Chemistry and WestCHEM, University of Glasgow, Glasgow G12 8QQ, United Kingdom
b School of Chemistry and EastCHEM, University of St Andrews, St Andrews KY16 9ST, United Kingdom
Abstract
The sidebands observed in the magic angle spinning (MAS) NMR spectrum of a spin I = 1 2H nucleus may be very strongly broadened if motion is present in the solid. The broadening arises from interference between the line-narrowing effects of MAS and the dynamics-driven reorientation of the 2H quadrupole tensor. Here, we show that this motional broadening is absent or much reduced in the corresponding double-quantum MAS NMR spectrum. This observation suggests a new approach to studying dynamics in solids and examples are drawn from 2H MAS NMR of oxalic acid dihydrate, sodium tetrathionate dihydrate, and the synthetic polymer PMMA.
Monday, May 22, 2006
Hiyam's Journal update
Chemistry - A European Journal Volume: 12, Issue: 10
14N NMR and Two-Dimensional Suspension 1H and 13C HRMAS NMR Spectroscopy of Ionic Liquids Immobilized on Silica
Brenna, Stefano; Posset, Tobias; Furrer, Julien; Blümel, Janet pp. 2880 - 2888
14N NMR and Two-Dimensional Suspension 1H and 13C HRMAS NMR Spectroscopy of Ionic Liquids Immobilized on Silica
Brenna, Stefano; Posset, Tobias; Furrer, Julien; Blümel, Janet pp. 2880 - 2888
Chemistry - A European Journal Volume: 12, Issue: 14
Orientational Effect of Aryl Groups on 77Se NMR Chemical Shifts: Experimental and Theoretical Investigations
Orientational Effect of Aryl Groups on 77Se NMR Chemical Shifts: Experimental and Theoretical Investigations
Nakanishi, Waro; Hayashi, Satoko; Shimizu, Daisuke; Hada, Masahiko pp. 3829 - 3846
Friday, May 19, 2006
Inorganic Chemistry:Investigation of Tetrahedral Mixed-Metal Carbonyl Clusters by Two-Dimensional 59Co COSY and DQFCOSY NMR Experiments
Inorg. Chem., 45 (8), 3378 -3383, 2006. 10.1021/ic051544a S0020-1669(05)01544-2 Web
Investigation of Tetrahedral Mixed-Metal Carbonyl Clusters by Two-Dimensional 59Co COSY and DQFCOSY NMR Experiments
Pierre Kempgens, Karim Elbayed, Jésus Raya, Pierre Granger, Jacky Rosé, and Pierre Braunstein*
Laboratoire de RMN de la Matière Condensée (UMR 7177 CNRS), Université Louis Pasteur, Institut de Chimie, BP 296, F-67008 Strasbourg Cedex, France, and Laboratoire de Chimie de Coordination (UMR 7177 CNRS), Université Louis Pasteur, 4 rue Blaise Pascal, F-67070 Strasbourg Cedex, France
Abstract: Two-dimensional (2D) 59Co correlation spectroscopy (COSY)/double-quantum-filtered (DQF)COSY experiments are reported for three tetrahedral mixed-metal clusters HFeCo3(CO)11L with L = PPh3, P(OMe)3, and PCy3 (Cy = cyclohexyl) in which the L-substituted Co center is chemically different from the other two. The 2D 59Co COSY and DQFCOSY NMR spectra of these clusters in solution prove the existence of a scalar coupling constant between the 59Co nuclei. To determine this value for each cluster, 2D 59Co COSY and DQFCOSY NMR spectra have been simulated by numerical density-matrix calculations. The predicted spectra mimic well the features of the experimental spectra if a scalar coupling is introduced between the Co nuclei. It was initially observed that the scalar coupling constants between the Co nuclei obtained from the 2D COSY and DQFCOSY NMR spectra differed significantly. In contrast to the 2D COSY spectra, the diagonal and cross peaks are of comparable intensity in the 2D DQFCOSY spectra, which leads to a considerable increase in the accuracy of the determination of the scalar coupling constant.
Investigation of Tetrahedral Mixed-Metal Carbonyl Clusters by Two-Dimensional 59Co COSY and DQFCOSY NMR Experiments
Pierre Kempgens, Karim Elbayed, Jésus Raya, Pierre Granger, Jacky Rosé, and Pierre Braunstein*
Laboratoire de RMN de la Matière Condensée (UMR 7177 CNRS), Université Louis Pasteur, Institut de Chimie, BP 296, F-67008 Strasbourg Cedex, France, and Laboratoire de Chimie de Coordination (UMR 7177 CNRS), Université Louis Pasteur, 4 rue Blaise Pascal, F-67070 Strasbourg Cedex, France
Abstract: Two-dimensional (2D) 59Co correlation spectroscopy (COSY)/double-quantum-filtered (DQF)COSY experiments are reported for three tetrahedral mixed-metal clusters HFeCo3(CO)11L with L = PPh3, P(OMe)3, and PCy3 (Cy = cyclohexyl) in which the L-substituted Co center is chemically different from the other two. The 2D 59Co COSY and DQFCOSY NMR spectra of these clusters in solution prove the existence of a scalar coupling constant between the 59Co nuclei. To determine this value for each cluster, 2D 59Co COSY and DQFCOSY NMR spectra have been simulated by numerical density-matrix calculations. The predicted spectra mimic well the features of the experimental spectra if a scalar coupling is introduced between the Co nuclei. It was initially observed that the scalar coupling constants between the Co nuclei obtained from the 2D COSY and DQFCOSY NMR spectra differed significantly. In contrast to the 2D COSY spectra, the diagonal and cross peaks are of comparable intensity in the 2D DQFCOSY spectra, which leads to a considerable increase in the accuracy of the determination of the scalar coupling constant.
Inorganic Chemistry: 195Pt Chemical Shift Calculations
Comment: Joel, you should take a look
Inorg. Chem., 45 (8), 3316 -3324, 2006. 10.1021/ic052143y S0020-1669(05)02143-9 Web
Toward an Accurate Determination of 195Pt Chemical Shifts by Density Functional Computations: The Importance of Unspecific Solvent Effects and the Dependence of Pt Magnetic Shielding Constants on Structural Parameters
Mariusz Sterzel and Jochen Autschbach*
Department of Chemistry, State University of New York at Buffalo, Buffalo, New York 14260-3000
Abstract:
Density functional theory using the zero-order regular approximate two-component relativistic Hamiltonian has been applied to calculate the 195Pt chemical shifts for the complexes [PtCl6]2-, [PtCl4]2-, and [Pt2(NH3)2Cl2((CH3)3CCONH)2(CH2COCH3)]Cl. It is demonstrated that, in contrast to recent findings by other authors, platinum chemical shift calculations require not only a basis set beyond polarized triple- quality for the metal atom but also, in principle, the consideration of explicit solvent molecules in addition to a continuum model for the first two complexes. We find that the inclusion of direct solvent-solute interactions at the quantum mechanical level is important for obtaining reasonable results despite that fact that these solvent effects are rather nonspecific. The importance of solvent effects has also implications on how experimental data should be interpreted. Further, in contrast to several previous studies of heavy-metal NMR parameters, functionals beyond the local density approximation were required both in the geometry optimization and the NMR calculations to obtain reasonable agreement between the computed and experimental NMR data. This comes with the disadvantage, however, of increased Pt-ligand bond distances leading to less good agreement with experiment for structural data. A detailed analysis of the results for the two chloroplatinate complexes is presented. The same computational procedure has then been applied to the dinuclear Pt(III) complex. Chemical shifts have been calculated with respect to both [PtCl6]2- and [PtCl4]2- chosen as the NMR reference, yielding good agreement with experiment. The determination of preferred solvent locations around the complexes studied turned out to be important for reproducing experimental data.
Inorg. Chem., 45 (8), 3316 -3324, 2006. 10.1021/ic052143y S0020-1669(05)02143-9 Web
Toward an Accurate Determination of 195Pt Chemical Shifts by Density Functional Computations: The Importance of Unspecific Solvent Effects and the Dependence of Pt Magnetic Shielding Constants on Structural Parameters
Mariusz Sterzel and Jochen Autschbach*
Department of Chemistry, State University of New York at Buffalo, Buffalo, New York 14260-3000
Abstract:
Density functional theory using the zero-order regular approximate two-component relativistic Hamiltonian has been applied to calculate the 195Pt chemical shifts for the complexes [PtCl6]2-, [PtCl4]2-, and [Pt2(NH3)2Cl2((CH3)3CCONH)2(CH2COCH3)]Cl. It is demonstrated that, in contrast to recent findings by other authors, platinum chemical shift calculations require not only a basis set beyond polarized triple- quality for the metal atom but also, in principle, the consideration of explicit solvent molecules in addition to a continuum model for the first two complexes. We find that the inclusion of direct solvent-solute interactions at the quantum mechanical level is important for obtaining reasonable results despite that fact that these solvent effects are rather nonspecific. The importance of solvent effects has also implications on how experimental data should be interpreted. Further, in contrast to several previous studies of heavy-metal NMR parameters, functionals beyond the local density approximation were required both in the geometry optimization and the NMR calculations to obtain reasonable agreement between the computed and experimental NMR data. This comes with the disadvantage, however, of increased Pt-ligand bond distances leading to less good agreement with experiment for structural data. A detailed analysis of the results for the two chloroplatinate complexes is presented. The same computational procedure has then been applied to the dinuclear Pt(III) complex. Chemical shifts have been calculated with respect to both [PtCl6]2- and [PtCl4]2- chosen as the NMR reference, yielding good agreement with experiment. The determination of preferred solvent locations around the complexes studied turned out to be important for reproducing experimental data.
Dalton Transactions:Silver nitrate in silver zeolite A: three-dimensional incommensurate guest ordering in a zeolite framework
Comment: Looks like it could be similar to some of Hiyam's samples
Dalton Transactions, 2006, 2368 - 2373DOI: 10.1039/b517094j
Silver nitrate in silver zeolite A: three-dimensional incommensurate guest ordering in a zeolite framework
M. Viertelhaus, A. E. Taylor, L. Kloo, I. Gameson and P. A. Anderson
Abstract: We report the results of a detailed examination of the occlusion of silver nitrate in silver zeolite A (AgA). The superlattice reported to occur in (AgNO3)9–AgA was found to melt at between 80 and 100 °C on heating and reappear when the sample was cooled down to 80 °C. Annealing in this temperature range and rigorous exclusion of water produced an enhancement of the superlattice peaks, which results from ordering of the contents of the zeolite cages. Peaks assigned to the superlattice were indexed with the tetragonal lattice parameters a = 17.440(5) and c = 12.398(4) Å and proposed space group P4/nmm. The sharp peaks representing the lattice of the framework (a = 12.3711(5) Å, Pmm) remained largely unaffected by the guest in this compound, which was found to exhibit strong negative thermal expansion. The host and guest lattices are incommensurate with the tetragonal guest lattice being slightly larger than the cubic host in the c-direction and slightly smaller in the a- and b-directions
Dalton Transactions, 2006, 2368 - 2373DOI: 10.1039/b517094j
Silver nitrate in silver zeolite A: three-dimensional incommensurate guest ordering in a zeolite framework
M. Viertelhaus, A. E. Taylor, L. Kloo, I. Gameson and P. A. Anderson
Abstract: We report the results of a detailed examination of the occlusion of silver nitrate in silver zeolite A (AgA). The superlattice reported to occur in (AgNO3)9–AgA was found to melt at between 80 and 100 °C on heating and reappear when the sample was cooled down to 80 °C. Annealing in this temperature range and rigorous exclusion of water produced an enhancement of the superlattice peaks, which results from ordering of the contents of the zeolite cages. Peaks assigned to the superlattice were indexed with the tetragonal lattice parameters a = 17.440(5) and c = 12.398(4) Å and proposed space group P4/nmm. The sharp peaks representing the lattice of the framework (a = 12.3711(5) Å, Pmm) remained largely unaffected by the guest in this compound, which was found to exhibit strong negative thermal expansion. The host and guest lattices are incommensurate with the tetragonal guest lattice being slightly larger than the cubic host in the c-direction and slightly smaller in the a- and b-directions
Chemisty of Materials:Platinum Nanoparticle Interaction with Chemically Modified Highly Oriented Pyrolytic Graphite Surfaces
Comment: 195Pt NMR application?
Chem. Mater., 18 (7), 1811 -1816, 2006. 10.1021/cm052453e S0897-4756(05)02453-1 Web
Platinum Nanoparticle Interaction with Chemically Modified Highly Oriented Pyrolytic Graphite Surfaces
De-Quan Yang and Edward Sacher*
Regroupement Québécois de Matériaux de Pointe, Département de Génie Physique, École Polytechnique, C.P. 6079, succursale Centre-Ville, Montréal, Québec H3C 3A7, Canada
Abstract:
Ar, N2, O2, and H2O radio frequency (13.56 MHz) plasma treatments have been carried out on insulator-supported samples of highly oriented pyrolytic graphite (HOPG). The HOPG surface was found to be minimally damaged by all the plasma treatments, which break C-C bonds and form free radicals in the outermost few layers. These free radicals subsequently react, on exposure to air, forming various oxidation products; both XPS and photoacoustic FTIR indicate that these include C-OH, C=O, and -COOH. Pt was deposited onto these samples by evaporation, forming nanoparticles. After plasma treatment, the dimensions of the Pt nanoparticles were ~3.5-4 nm, compared to ~6 nm on untreated HOPG, and they no longer diffused laterally; this indicates enhanced interfacial adhesion for all these plasma treatments. The occurrence of oxidation products strongly suggests that the increased Pt nanoparticle adhesion to the treated HOPG surface is due to their presence.
Chem. Mater., 18 (7), 1811 -1816, 2006. 10.1021/cm052453e S0897-4756(05)02453-1 Web
Platinum Nanoparticle Interaction with Chemically Modified Highly Oriented Pyrolytic Graphite Surfaces
De-Quan Yang and Edward Sacher*
Regroupement Québécois de Matériaux de Pointe, Département de Génie Physique, École Polytechnique, C.P. 6079, succursale Centre-Ville, Montréal, Québec H3C 3A7, Canada
Abstract:
Ar, N2, O2, and H2O radio frequency (13.56 MHz) plasma treatments have been carried out on insulator-supported samples of highly oriented pyrolytic graphite (HOPG). The HOPG surface was found to be minimally damaged by all the plasma treatments, which break C-C bonds and form free radicals in the outermost few layers. These free radicals subsequently react, on exposure to air, forming various oxidation products; both XPS and photoacoustic FTIR indicate that these include C-OH, C=O, and -COOH. Pt was deposited onto these samples by evaporation, forming nanoparticles. After plasma treatment, the dimensions of the Pt nanoparticles were ~3.5-4 nm, compared to ~6 nm on untreated HOPG, and they no longer diffused laterally; this indicates enhanced interfacial adhesion for all these plasma treatments. The occurrence of oxidation products strongly suggests that the increased Pt nanoparticle adhesion to the treated HOPG surface is due to their presence.
Chemistry of Materials:Pt Nanoparticle Binding on Functionalized Multiwalled Carbon Nanotubes
Comment: Application of 195Pt NMR?? Looks like an interesting system to characterize.
Chem. Mater., 18 (7), 1780 -1788, 2006. 10.1021/cm0518978 S0897-4756(05)01897-1 Web
Pt Nanoparticle Binding on Functionalized Multiwalled Carbon Nanotubes
Robert V. Hull, Liang Li, Yangchuan Xing, and Charles C. Chusuei*
Departments of Chemistry and Chemical and Biological Engineering, University of Missouri-Rolla, Rolla, Missouri 65409-0010
Abstract:
To create new catalyst materials for fuel cell applications, multiwalled carbon nanotubes (CNTs) were functionalized with -C=O, -C-O-C-, -COO-, and -C-OH groups using a sonochemical treatment method under acidic aqueous solution (HNO3/H2SO4) conditions to make them amenable to deposition of highly dispersed, ~4 nm diameter Pt nanoparticles. The Pt-CNT interface was probed with X-ray photoelectron spectroscopy (XPS), extended X-ray absorption fine structure spectroscopy (EXAFS), and Raman and attenuated total reflection infrared (ATR-IR) spectroscopies to elucidate the nature of the Pt cluster-CNT surface binding. The degree of disorder of the sp3-hybridized C from the CNTs, as measured by the Raman D-to-G integrated peak area ratios, increased with the degree of surface oxidation of the CNTs. EXAFS of the Pt LIII edge showed Pt coordination with oxygen (in the form of PtOx) at the outermost perimeter of the Pt clusters while the majority of the bulk, as shown by the XPS Pt 4f core level, was in the metallic form. Infrared measurements showed that the carbonyl C=O stretching at 1700 cm-1 red shifted to ~1550 cm-1 following Pt cluster deposition. In addition, changes in the C-O structural features at ~1030 and 1150 cm-1 were observed, indicative of Pt cluster binding with the ionic form of carboxylate, COO(Pt), or ester-like, C(=O)CO(Pt), O atoms.
Chem. Mater., 18 (7), 1780 -1788, 2006. 10.1021/cm0518978 S0897-4756(05)01897-1 Web
Pt Nanoparticle Binding on Functionalized Multiwalled Carbon Nanotubes
Robert V. Hull, Liang Li, Yangchuan Xing, and Charles C. Chusuei*
Departments of Chemistry and Chemical and Biological Engineering, University of Missouri-Rolla, Rolla, Missouri 65409-0010
Abstract:
To create new catalyst materials for fuel cell applications, multiwalled carbon nanotubes (CNTs) were functionalized with -C=O, -C-O-C-, -COO-, and -C-OH groups using a sonochemical treatment method under acidic aqueous solution (HNO3/H2SO4) conditions to make them amenable to deposition of highly dispersed, ~4 nm diameter Pt nanoparticles. The Pt-CNT interface was probed with X-ray photoelectron spectroscopy (XPS), extended X-ray absorption fine structure spectroscopy (EXAFS), and Raman and attenuated total reflection infrared (ATR-IR) spectroscopies to elucidate the nature of the Pt cluster-CNT surface binding. The degree of disorder of the sp3-hybridized C from the CNTs, as measured by the Raman D-to-G integrated peak area ratios, increased with the degree of surface oxidation of the CNTs. EXAFS of the Pt LIII edge showed Pt coordination with oxygen (in the form of PtOx) at the outermost perimeter of the Pt clusters while the majority of the bulk, as shown by the XPS Pt 4f core level, was in the metallic form. Infrared measurements showed that the carbonyl C=O stretching at 1700 cm-1 red shifted to ~1550 cm-1 following Pt cluster deposition. In addition, changes in the C-O structural features at ~1030 and 1150 cm-1 were observed, indicative of Pt cluster binding with the ionic form of carboxylate, COO(Pt), or ester-like, C(=O)CO(Pt), O atoms.
Chemical Physics: Detection of Chirality via Solution NMR.
Comment: Detection of chirality via solution NMR. Could have many applications.
Chemical Physics
Vol: 324 Issue: 1, May 9, 2006
pp: 111-116
Direct chiral discrimination in NMR spectroscopy
A.D. Buckingham a, , P. Fischer b, a Department of Chemistry, Cambridge University, Cambridge CB2 1EW, UKb The Rowland Institute at Harvard, Harvard University, Cambridge, MA 02142, USAReceived 30 June 2005; accepted 6 October 2005
Abstract:
Conventional nuclear magnetic resonance spectroscopy is unable to distinguish between the two mirror-image forms (enantiomers) of a chiral molecule. This is because the NMR spectrum is determined by the chemical shifts and spin–spin coupling constants which – in the absence of a chiral solvent – are identical for the two enantiomers. We discuss how chirality may nevertheless be directly detected in liquid-state NMR spectroscopy: In a chiral molecule, the rotating nuclear magnetic moment induces an electric dipole moment in the direction perpendicular to itself and to the permanent magnetic field of the spectrometer. We present computations of the precessing electric polarization following a π/2 pulse. Our estimates indicate that the electric polarization should be detectable in favourable cases. We also predict that application of an electrostatic field induces a chirally sensitive magnetization oscillating in the direction of the permanent magnetic field. We show that the electric-field-perturbed chemical shift tensor, the nuclear magnetic shielding polarizability, underlies these chiral NMR effects.
Chemical Physics
Vol: 324 Issue: 1, May 9, 2006
pp: 111-116
Direct chiral discrimination in NMR spectroscopy
A.D. Buckingham a, , P. Fischer b, a Department of Chemistry, Cambridge University, Cambridge CB2 1EW, UKb The Rowland Institute at Harvard, Harvard University, Cambridge, MA 02142, USAReceived 30 June 2005; accepted 6 October 2005
Abstract:
Conventional nuclear magnetic resonance spectroscopy is unable to distinguish between the two mirror-image forms (enantiomers) of a chiral molecule. This is because the NMR spectrum is determined by the chemical shifts and spin–spin coupling constants which – in the absence of a chiral solvent – are identical for the two enantiomers. We discuss how chirality may nevertheless be directly detected in liquid-state NMR spectroscopy: In a chiral molecule, the rotating nuclear magnetic moment induces an electric dipole moment in the direction perpendicular to itself and to the permanent magnetic field of the spectrometer. We present computations of the precessing electric polarization following a π/2 pulse. Our estimates indicate that the electric polarization should be detectable in favourable cases. We also predict that application of an electrostatic field induces a chirally sensitive magnetization oscillating in the direction of the permanent magnetic field. We show that the electric-field-perturbed chemical shift tensor, the nuclear magnetic shielding polarizability, underlies these chiral NMR effects.
Chemical Physics: 17O and 1H shielding calculations
Comment: Cory, looks like someone beat you to the discovery of 17O CSA in water.
Volume: 323, Issue: 2-3 April 21, 2006
pp. 218-230
On the calculations of the nuclear shielding constants in small water clusters
Hubert Cybulski, Joanna Sadlej , Department of Chemistry, University of Warsaw, Pasteura 1,
Abstract
The comparative study of the calculated ab initio 17 O and 1 H shielding constants for water clusters (H2 O)n, n = 2–6, 12 and 17, is presented. The comparison of different methods and the convergence of a basis set size were analyzed to enable the choice of the most efficient method of calculations. DFT(B3LYP)/aug-cc-pCVDZ calculations of shielding constants were performed for all clusters under the study. The correlation of the changes in the 1 H shielding constants on intra- and intermolecular distances was observed. The changes of the 17 O shielding constants depend strongly on the cluster size and its hydrogen-bonding topology. The largest shift (−76.2 ppm) was found for the central oxygen atom surrounded by two hydration shells in the largest water cluster, (H2 O)17. The interaction-induced changes in the 17 O shielding constants are found to be non-additive.
Volume: 323, Issue: 2-3 April 21, 2006
pp. 218-230
On the calculations of the nuclear shielding constants in small water clusters
Hubert Cybulski, Joanna Sadlej , Department of Chemistry, University of Warsaw, Pasteura 1,
Abstract
The comparative study of the calculated ab initio 17 O and 1 H shielding constants for water clusters (H2 O)n, n = 2–6, 12 and 17, is presented. The comparison of different methods and the convergence of a basis set size were analyzed to enable the choice of the most efficient method of calculations. DFT(B3LYP)/aug-cc-pCVDZ calculations of shielding constants were performed for all clusters under the study. The correlation of the changes in the 1 H shielding constants on intra- and intermolecular distances was observed. The changes of the 17 O shielding constants depend strongly on the cluster size and its hydrogen-bonding topology. The largest shift (−76.2 ppm) was found for the central oxygen atom surrounded by two hydration shells in the largest water cluster, (H2 O)17. The interaction-induced changes in the 17 O shielding constants are found to be non-additive.
Thursday, May 18, 2006
JPCB, Rienstra, 15N CSA in proteins; 3D MAS NMR
J. Phys. Chem. B, ASAP Article 10.1021/jp060507h S1520-6106(06)00507-4
Determinations of 15N Chemical Shift Anisotropy Magnitudes in a Uniformly 15N,13C-Labeled Microcrystalline Protein by Three-Dimensional Magic-Angle Spinning Nuclear Magnetic Resonance Spectroscopy
Benjamin J. Wylie, W. Trent Franks, and Chad M. Rienstra*
Department of Chemistry, Department of Biochemistry, and Center for Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, Illinois 61801
Abstract:
Amide 15N chemical shift anisotropy (CSA) tensors provide quantitative insight into protein structure and dynamics. Experimental determinations of 15N CSA tensors in biologically relevant molecules have typically been performed by NMR relaxation studies in solution, goniometric analysis of single-crystal spectra, or slow magic-angle spinning (MAS) NMR experiments of microcrystalline samples. Here we present measurements of 15N CSA tensor magnitudes in a protein of known structure by three-dimensional MAS solid-state NMR. Isotropic 15N, 13C, and 13C' chemical shifts in two dimensions resolve site-specific backbone amide recoupled CSA line shapes in the third dimension. Application of the experiments to the 56-residue 1 immunoglobulin binding domain of protein G (GB1) enabled 91 independent determinations of 15N tensors at 51 of the 55 backbone amide sites, for which 15N-13C and/or 15N-13C' cross-peaks were resolved in the two-dimensional experiment. For 37 15N signals, both intra- and interresidue correlations were resolved, enabling direct comparison of two experimental data sets to enhance measurement precision. Systematic variations between -sheet and -helix residues are observed; the average value for the anisotropy parameter, ( = zz - iso), for -helical residues is 6 ppm greater than that for the -sheet residues. The results show a variation in of 15N amide backbone sites between -77 and -115 ppm, with an average value of -103.5 ppm. Some sites (e.g., G41) display smaller anisotropy due to backbone dynamics. In contrast, we observe an unusually large 15N tensor for K50, a residue that has an atypical, positive value for the backbone torsion angle. To our knowledge, this is the most complete experimental analysis of 15N CSA magnitude to date in a solid protein. The availability of previous high-resolution crystal and solution NMR structures, as well as detailed solid-state NMR studies, will enhance the value of these measurements as a benchmark for the development of ab initio calculations of amide 15N shielding tensor magnitudes.
Determinations of 15N Chemical Shift Anisotropy Magnitudes in a Uniformly 15N,13C-Labeled Microcrystalline Protein by Three-Dimensional Magic-Angle Spinning Nuclear Magnetic Resonance Spectroscopy
Benjamin J. Wylie, W. Trent Franks, and Chad M. Rienstra*
Department of Chemistry, Department of Biochemistry, and Center for Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, Illinois 61801
Abstract:
Amide 15N chemical shift anisotropy (CSA) tensors provide quantitative insight into protein structure and dynamics. Experimental determinations of 15N CSA tensors in biologically relevant molecules have typically been performed by NMR relaxation studies in solution, goniometric analysis of single-crystal spectra, or slow magic-angle spinning (MAS) NMR experiments of microcrystalline samples. Here we present measurements of 15N CSA tensor magnitudes in a protein of known structure by three-dimensional MAS solid-state NMR. Isotropic 15N, 13C, and 13C' chemical shifts in two dimensions resolve site-specific backbone amide recoupled CSA line shapes in the third dimension. Application of the experiments to the 56-residue 1 immunoglobulin binding domain of protein G (GB1) enabled 91 independent determinations of 15N tensors at 51 of the 55 backbone amide sites, for which 15N-13C and/or 15N-13C' cross-peaks were resolved in the two-dimensional experiment. For 37 15N signals, both intra- and interresidue correlations were resolved, enabling direct comparison of two experimental data sets to enhance measurement precision. Systematic variations between -sheet and -helix residues are observed; the average value for the anisotropy parameter, ( = zz - iso), for -helical residues is 6 ppm greater than that for the -sheet residues. The results show a variation in of 15N amide backbone sites between -77 and -115 ppm, with an average value of -103.5 ppm. Some sites (e.g., G41) display smaller anisotropy due to backbone dynamics. In contrast, we observe an unusually large 15N tensor for K50, a residue that has an atypical, positive value for the backbone torsion angle. To our knowledge, this is the most complete experimental analysis of 15N CSA magnitude to date in a solid protein. The availability of previous high-resolution crystal and solution NMR structures, as well as detailed solid-state NMR studies, will enhance the value of these measurements as a benchmark for the development of ab initio calculations of amide 15N shielding tensor magnitudes.
Wednesday, May 17, 2006
Cory's Journals - April 2006
April 2006 articles from the journals that I am responsible for will be placed in their usual folder by the end of today
Tuesday, May 16, 2006
Joel: JSSC Update
Cory, any interest?
NMR Study of the Order-Disorder Phase Transition of KHCO3 and KDCO3 Single Crystals.
A.R. Lim and S.Y. Jeong
JSolidStateChem (2006) 179, 1009.
Abstract:
KHCO3 and its deuterated analogue KDCO3 are typical materials that undergo order–disorder phase transitions at 318 and 353 K, respectively. The spin–lattice relaxation times, T1, spin–spin relaxation times, T2, and the number of resonance lines for the 1H, 2D, and 39K nuclei of these crystals were investigated using NMR spectrometer. These materials are known to exhibit anomalous decreases in T1 near TC, which have been attributed to a structural phase transition. Additionally, changes in the symmetry of the (HCO3)22- (or (DCO3)22-) dimers in these materials are associated with large changes in T1, T2, and the number of resonance lines. Here we found that the resonance lines for 1H, 2D, and 39K nuclei decrease in number as the temperature is increased up to TC, indicating that the orientations of the (HCO3)22- (or (DCO3)22- ) dimers and the environments of the K ions change at TC. Moreover, based on number of resonance lines, the results further indicate that the (HCO3)22- (or (DCO3)22- ) dimers reorientate to approximately parallel to the directions of the hydrogen bonds (or deuteron bonds) and the direction of the a-axis. The transitions at 318 and 345K of the two crystals are of the order–disorder type. The present results therefore indicate that the orientations of the (HCO3)22- and (DCO3)22- dimers and the environment of the K ion play a significant role in these phase transitions.
Rob, Cu(I)-I chains
Synthesis, structure and optical limiting effect of a novel inorganic-organic hybrid polymer containing mixed chains of copper(I)/iodine
H.H. Li et al.
JSolidStateChem (2006) 179, 1415.
Abstract:
In this paper, treatment of N-ethyl-benzo[f]quinolium (ebq) iodide and CuI with excess KI afforded an unusual coordination polymer [(ebq)2(Cu3I4)(CuI2)]n (1). 1 crystallizes in tetragonal system, space group P4(2)bc with cell parameters of a = 23.2040(6)A, c = 6.7393(3)A, V = 3628.6(2)A3, Z = 8, Dc= 2.622 g/c3, R1 = 0.0447 and wR2 = 0.0974. A highly interesting feature of 1 is its presence of mixed types of chains [(Cu3I4)-n and (CuI2)-n chain] in one crystal lattice based on supramolecular self-assembly directed by cations. The in.nite chains (Cu3I4)n and (CuI2)n in 1 could be described as the edge-sharing arrangement of CuI4 tetrahedron. Furthermore, IR, EA, UV–Vis, thermal analysis and optical limiting measurements were adopted to characterize polymer 1. The optical limiting experiment shows that the present polymer exhibits a large optical limiting capacity.
NMR Study of the Order-Disorder Phase Transition of KHCO3 and KDCO3 Single Crystals.
A.R. Lim and S.Y. Jeong
JSolidStateChem (2006) 179, 1009.
Abstract:
KHCO3 and its deuterated analogue KDCO3 are typical materials that undergo order–disorder phase transitions at 318 and 353 K, respectively. The spin–lattice relaxation times, T1, spin–spin relaxation times, T2, and the number of resonance lines for the 1H, 2D, and 39K nuclei of these crystals were investigated using NMR spectrometer. These materials are known to exhibit anomalous decreases in T1 near TC, which have been attributed to a structural phase transition. Additionally, changes in the symmetry of the (HCO3)22- (or (DCO3)22-) dimers in these materials are associated with large changes in T1, T2, and the number of resonance lines. Here we found that the resonance lines for 1H, 2D, and 39K nuclei decrease in number as the temperature is increased up to TC, indicating that the orientations of the (HCO3)22- (or (DCO3)22- ) dimers and the environments of the K ions change at TC. Moreover, based on number of resonance lines, the results further indicate that the (HCO3)22- (or (DCO3)22- ) dimers reorientate to approximately parallel to the directions of the hydrogen bonds (or deuteron bonds) and the direction of the a-axis. The transitions at 318 and 345K of the two crystals are of the order–disorder type. The present results therefore indicate that the orientations of the (HCO3)22- and (DCO3)22- dimers and the environment of the K ion play a significant role in these phase transitions.
Rob, Cu(I)-I chains
Synthesis, structure and optical limiting effect of a novel inorganic-organic hybrid polymer containing mixed chains of copper(I)/iodine
H.H. Li et al.
JSolidStateChem (2006) 179, 1415.
Abstract:
In this paper, treatment of N-ethyl-benzo[f]quinolium (ebq) iodide and CuI with excess KI afforded an unusual coordination polymer [(ebq)2(Cu3I4)(CuI2)]n (1). 1 crystallizes in tetragonal system, space group P4(2)bc with cell parameters of a = 23.2040(6)A, c = 6.7393(3)A, V = 3628.6(2)A3, Z = 8, Dc= 2.622 g/c3, R1 = 0.0447 and wR2 = 0.0974. A highly interesting feature of 1 is its presence of mixed types of chains [(Cu3I4)-n and (CuI2)-n chain] in one crystal lattice based on supramolecular self-assembly directed by cations. The in.nite chains (Cu3I4)n and (CuI2)n in 1 could be described as the edge-sharing arrangement of CuI4 tetrahedron. Furthermore, IR, EA, UV–Vis, thermal analysis and optical limiting measurements were adopted to characterize polymer 1. The optical limiting experiment shows that the present polymer exhibits a large optical limiting capacity.
Joel: Advan. Mat. Update
Andy
Luminescence Spectra and Dynamics of Mn-Doped CdS Core/Shell Nanocrystals.
A. Ishtzumt and Y. Kanemitsu
Advan. Mat. (2006)18, 1083.
Summary:
The overcoating of CdS:Mn nanocrystals by a thin shell of non-doped CdS or ZnS results in remarkably improved photoluminescence properties. The CdS:Mn/CdS and CdS:Mn/ZnS nanocrystals presented in this work (see figure) show a much more efficient Mn-related photoluminescence than the non-coated nanocrystals. These nanocrystals are expected to find applications in flat-panel displays, high-density optical data storage materials, and fluorescence labels for biological imaging
Bryan
Growing Metal Nanoparticles by Enzymes
I. Willner, R. Baron, B. Willner.
Advan. Mat. (2006)18, 1109.
Abstract:
Enzymes act as catalysts for the growth of metallic nanoparticles (NPs). The enzymemediated growth of metallic NPs provides a general means to follow biocatalyzed transformations, and to develop optical sensors for different substrates such as glucose, L-DOPA, alcohols, lactate or nerve gas analogs. Enzymes modified with Au NPs act as biocatalysts for the fabrication of metallic nanowires. The dip-pen nanolithography of NP-functionalized enzymes on Si surfaces yields biocatalytic templates that enable the orthogonal evolution of nanowires consisting of different metals.
Andre, Joel
Molecular Materials by Self-Assembly of Porphyrins, Phthalocyanince, and Perylenes
J.A.A.W. Elemans, R. van Hameren, J.M. Nolte, A.E. Rowan.
Advan. Mat. (2006)18, 1251.
Abstract:
Porphyrins, phthalocyanines, and perylenes are compounds with great potential for serving as components of molecular materials that possess unique electronic, magnetic and photophysical properties. In general, a highly specific communication between a large number of these chromophores is required in order to express their function to a maximal level, and for this reason it is of importance to construct arrays in which the molecules are organized in well-defined geometries with respect to their neighbors. This review is an account of some recent efforts to construct highly ordered assemblies of porphyrins, phthalocyanines, and perylenes by means of self-assembly in solution and on surfaces, and by attaching them to polymeric scaffolds.
Rob, Interesting Cu(I) complex...
Highly Luminescent Cu(I) Complexes for Light-Emitting Electrochemical Cells.
N. Armaroli, G. Accorst, M. Holler, O. Moudam, J.F. Nierengarten, Z. Zhou, R.T. Wegh, R. Welter.
Advan. Mat. (2006)18, 1313.
Summary:
Highly luminescent CuI complexes with one phenanthroline and one (bis[2-(diphenylphosphino)phenyl]ether) ligand show an emission quantum yield of up to 28 % upon deactivation of the metal-to-ligand charge-transfer excited states; their X-ray crystal structure shows a distorted tetrahedral geometry (see figure and inside cover). One of these compounds is used as an active material in a light-emitting electrochemical cell; it exhibits an efficiency similar to that of RuII-type complexes.
Luminescence Spectra and Dynamics of Mn-Doped CdS Core/Shell Nanocrystals.
A. Ishtzumt and Y. Kanemitsu
Advan. Mat. (2006)18, 1083.
Summary:
The overcoating of CdS:Mn nanocrystals by a thin shell of non-doped CdS or ZnS results in remarkably improved photoluminescence properties. The CdS:Mn/CdS and CdS:Mn/ZnS nanocrystals presented in this work (see figure) show a much more efficient Mn-related photoluminescence than the non-coated nanocrystals. These nanocrystals are expected to find applications in flat-panel displays, high-density optical data storage materials, and fluorescence labels for biological imaging
Bryan
Growing Metal Nanoparticles by Enzymes
I. Willner, R. Baron, B. Willner.
Advan. Mat. (2006)18, 1109.
Abstract:
Enzymes act as catalysts for the growth of metallic nanoparticles (NPs). The enzymemediated growth of metallic NPs provides a general means to follow biocatalyzed transformations, and to develop optical sensors for different substrates such as glucose, L-DOPA, alcohols, lactate or nerve gas analogs. Enzymes modified with Au NPs act as biocatalysts for the fabrication of metallic nanowires. The dip-pen nanolithography of NP-functionalized enzymes on Si surfaces yields biocatalytic templates that enable the orthogonal evolution of nanowires consisting of different metals.
Andre, Joel
Molecular Materials by Self-Assembly of Porphyrins, Phthalocyanince, and Perylenes
J.A.A.W. Elemans, R. van Hameren, J.M. Nolte, A.E. Rowan.
Advan. Mat. (2006)18, 1251.
Abstract:
Porphyrins, phthalocyanines, and perylenes are compounds with great potential for serving as components of molecular materials that possess unique electronic, magnetic and photophysical properties. In general, a highly specific communication between a large number of these chromophores is required in order to express their function to a maximal level, and for this reason it is of importance to construct arrays in which the molecules are organized in well-defined geometries with respect to their neighbors. This review is an account of some recent efforts to construct highly ordered assemblies of porphyrins, phthalocyanines, and perylenes by means of self-assembly in solution and on surfaces, and by attaching them to polymeric scaffolds.
Rob, Interesting Cu(I) complex...
Highly Luminescent Cu(I) Complexes for Light-Emitting Electrochemical Cells.
N. Armaroli, G. Accorst, M. Holler, O. Moudam, J.F. Nierengarten, Z. Zhou, R.T. Wegh, R. Welter.
Advan. Mat. (2006)18, 1313.
Summary:
Highly luminescent CuI complexes with one phenanthroline and one (bis[2-(diphenylphosphino)phenyl]ether) ligand show an emission quantum yield of up to 28 % upon deactivation of the metal-to-ligand charge-transfer excited states; their X-ray crystal structure shows a distorted tetrahedral geometry (see figure and inside cover). One of these compounds is used as an active material in a light-emitting electrochemical cell; it exhibits an efficiency similar to that of RuII-type complexes.
Joel: JPCB Update
Hiyam
Analysis of Conformational Polymorphism in Pharmaceutical Solids Using Solid-State NMR and Electronic Structure Calculation
J.R. Smith, W. Xu, and D. Raftery
J.Phys.Chem.B (2006)110, 7766.
Abstract:
A detailed analysis of molecular structure in three polymorphic forms of 5-methyl-2-[(2-nitrophenyl)amino]- 3-thiophenecarbonitrile is made using a combination of multidimensional solid-state NMR (SSNMR) experiments and molecular modeling via electronic structure calculations. These compounds, collectively referred to as ROY because of their red, orange, and yellow colors, share a similar molecular structure with the exception of the dihedral angle between the phenyl and thiophene rings. The ROY materials make it possible to study the influence of nearly a single degree of freedom on the associated NMR spectra. Using the 2D PASS (Antzutkin et al. J. Magn. Reson. A 1995, 115, 7) experiment, spectral editing techniques, and DFT-based calculations of the local fields, an analysis is made of the sensitivity of all carbon and nitrogen sites to changing molecular conformation. Chemical shift and dipolar coupling information obtained from these experiments vary noticeably between forms and are subsequently used to quantitatively determine aspects of molecular structure in these materials, including the coplanar angle between the phenyl and thiophene rings. The influence of motion on the methyl and nitro chemical shifts is also investigated. The accuracy of the information obtained from local field analysis and the model structure calculation demonstrates the capabilities of SSNMR as a quantitative structural method.
Anyone interested in REDOR
Homonuclear and Heteronuclear NMR Studies of a Statherin Fragment Bound to Hydroxyapatite Crystals.
V. Raghunathan, J.M. Gibson, G. Goobes, J.M. Popham, E.A. Louie, P.S. Stayton, G.P. Drobny
J.Phys.Chem.B (2006)110, 9324.
Abstract:
Acidic proteins found in mineralized tissues act as nature's crystal engineers, where they play a key role in promoting or inhibiting the growth of minerals such as hydroxyapatite (HAP), Ca10(PO4)6(OH)2, the main mineral component of bone and teeth. Key to understanding the structural basis of protein-crystal recognition and protein control of hard tissue growth is the nature of interactions between the protein side chains and the crystal surface. In an earlier work we have measured the proximity of the lysine (K6) side chain in an SN-15 peptide fragment of the salivary protein statherin adsorbed to the Phosphorus-rich surface of HAP using solid-state NMR recoupling experiments. 15N{31P} rotational echo double resonance (REDOR) NMR data on the side-chain nitrogen in K6 gave rise to three different models of protein-surface interaction to explain the experimental data acquired. In this work we extend the analysis of the REDOR data by examining the
contribution of interactions between surface phosphorus atoms to the observed 15N REDOR decay. We performed 31P-31P recoupling experiments in HAP and (NH4)2HPO4 (DHP) to explore the nature of dipolar coupled 31P spin networks. These studies indicate that extensive networks of dipolar coupled 31P spins can be represented as stronger effective dipolar couplings, the existence of which must be included in the analysis of REDOR data. We carried out 15N{31P} REDOR in the case of DHP to determine how the size of the dephasing spin network influences the interpretation of the REDOR data. Although use of an extended 31P coupled spin network simulates the REDOR data well, a simplified 31P dephasing system composed of two spins with a larger dipolar coupling also simulates the REDOR data and only perturbs the heteronuclear couplings very slightly. The 31P-31P dipolar couplings between phosphorus nuclei in HAP can be replaced by an effective dipolar interaction of 600 Hz between two 31P spins. We incorporated this coupling and applied the above approach to reanalyze the 15N{31P} REDOR of the lysine side chain approaching the HAP surface and have refined the binding models proposed earlier. We obtain 15N-31P distances between 3.3 and 5 Å from these models that are indicative of the possibility of a lysine-phosphate hydrogen bond.
REDOR again
Short and Medium Range Order in Sodium Aluminophosphate Glasses: New Insights from High-resolution Dipolar Solid-State NMR Spectroscopy.
L. Zhang and H. Eckert
J.Phys.Chem.B (2006)110, 8946.
Abstract:
The structures of sodium aluminophosphate glasses prepared by both sol-gel as well as melt-cooling routes have been extensively characterized by high-resolution solid-state 23Na, 27Al, and 31P single and doubleresonance NMR techniques, including quantitative connectivity studies by 27Al T 31P and 23Na T 31P rotational echo double-resonance (REDOR) methods. Studies along four compositional lines, I: (AlPO4)x-(NaPO3)1-x, II: (Na2O)x-(AlPO4)1-x, III: (NaAlO2)x-(NaPO3)1-x, and IV: (Al2O3)x(NaPO3)1-x, reveal that the network structures of those glasses that are accessible by either preparation method are essentially identical. However, the significantly extended glass-forming ranges available by the sol-gel route facilitate exploration of the structure/composition relationships in more detail, revealing a number of interesting universal features throughout the whole glass system. Both short- and medium-range order appear to be controlled strongly by the O/P ratio of the glasses studied: Up to an O/P ratio of 3.5 (pyrophosphate composition), aluminum is predominantly six-coordinated and fully connected to phosphorus (Al(OP)6 sites). In the region 3.5 e O/P e 4.0, a dramatic structural transformation takes place, leading to the appearance of additional four- and fivecoordinated aluminum species whose second coordination spheres are also entirely dominated by phosphorus. The structure of glasses with an O/P ratio of precisely 4.0 (orthophosphate) is dominated by Al(OP)4 units. As the O/P ratio increases beyond 4.0, the average extent of Al-O-P connectivity is decreased significantly. Here, new types of five- and six-coordinated aluminum units, which are only weakly connected to ph osphorus, are formed, while the network modifier is attracted mainly by the phosphate units.
Analysis of Conformational Polymorphism in Pharmaceutical Solids Using Solid-State NMR and Electronic Structure Calculation
J.R. Smith, W. Xu, and D. Raftery
J.Phys.Chem.B (2006)110, 7766.
Abstract:
A detailed analysis of molecular structure in three polymorphic forms of 5-methyl-2-[(2-nitrophenyl)amino]- 3-thiophenecarbonitrile is made using a combination of multidimensional solid-state NMR (SSNMR) experiments and molecular modeling via electronic structure calculations. These compounds, collectively referred to as ROY because of their red, orange, and yellow colors, share a similar molecular structure with the exception of the dihedral angle between the phenyl and thiophene rings. The ROY materials make it possible to study the influence of nearly a single degree of freedom on the associated NMR spectra. Using the 2D PASS (Antzutkin et al. J. Magn. Reson. A 1995, 115, 7) experiment, spectral editing techniques, and DFT-based calculations of the local fields, an analysis is made of the sensitivity of all carbon and nitrogen sites to changing molecular conformation. Chemical shift and dipolar coupling information obtained from these experiments vary noticeably between forms and are subsequently used to quantitatively determine aspects of molecular structure in these materials, including the coplanar angle between the phenyl and thiophene rings. The influence of motion on the methyl and nitro chemical shifts is also investigated. The accuracy of the information obtained from local field analysis and the model structure calculation demonstrates the capabilities of SSNMR as a quantitative structural method.
Anyone interested in REDOR
Homonuclear and Heteronuclear NMR Studies of a Statherin Fragment Bound to Hydroxyapatite Crystals.
V. Raghunathan, J.M. Gibson, G. Goobes, J.M. Popham, E.A. Louie, P.S. Stayton, G.P. Drobny
J.Phys.Chem.B (2006)110, 9324.
Abstract:
Acidic proteins found in mineralized tissues act as nature's crystal engineers, where they play a key role in promoting or inhibiting the growth of minerals such as hydroxyapatite (HAP), Ca10(PO4)6(OH)2, the main mineral component of bone and teeth. Key to understanding the structural basis of protein-crystal recognition and protein control of hard tissue growth is the nature of interactions between the protein side chains and the crystal surface. In an earlier work we have measured the proximity of the lysine (K6) side chain in an SN-15 peptide fragment of the salivary protein statherin adsorbed to the Phosphorus-rich surface of HAP using solid-state NMR recoupling experiments. 15N{31P} rotational echo double resonance (REDOR) NMR data on the side-chain nitrogen in K6 gave rise to three different models of protein-surface interaction to explain the experimental data acquired. In this work we extend the analysis of the REDOR data by examining the
contribution of interactions between surface phosphorus atoms to the observed 15N REDOR decay. We performed 31P-31P recoupling experiments in HAP and (NH4)2HPO4 (DHP) to explore the nature of dipolar coupled 31P spin networks. These studies indicate that extensive networks of dipolar coupled 31P spins can be represented as stronger effective dipolar couplings, the existence of which must be included in the analysis of REDOR data. We carried out 15N{31P} REDOR in the case of DHP to determine how the size of the dephasing spin network influences the interpretation of the REDOR data. Although use of an extended 31P coupled spin network simulates the REDOR data well, a simplified 31P dephasing system composed of two spins with a larger dipolar coupling also simulates the REDOR data and only perturbs the heteronuclear couplings very slightly. The 31P-31P dipolar couplings between phosphorus nuclei in HAP can be replaced by an effective dipolar interaction of 600 Hz between two 31P spins. We incorporated this coupling and applied the above approach to reanalyze the 15N{31P} REDOR of the lysine side chain approaching the HAP surface and have refined the binding models proposed earlier. We obtain 15N-31P distances between 3.3 and 5 Å from these models that are indicative of the possibility of a lysine-phosphate hydrogen bond.
REDOR again
Short and Medium Range Order in Sodium Aluminophosphate Glasses: New Insights from High-resolution Dipolar Solid-State NMR Spectroscopy.
L. Zhang and H. Eckert
J.Phys.Chem.B (2006)110, 8946.
Abstract:
The structures of sodium aluminophosphate glasses prepared by both sol-gel as well as melt-cooling routes have been extensively characterized by high-resolution solid-state 23Na, 27Al, and 31P single and doubleresonance NMR techniques, including quantitative connectivity studies by 27Al T 31P and 23Na T 31P rotational echo double-resonance (REDOR) methods. Studies along four compositional lines, I: (AlPO4)x-(NaPO3)1-x, II: (Na2O)x-(AlPO4)1-x, III: (NaAlO2)x-(NaPO3)1-x, and IV: (Al2O3)x(NaPO3)1-x, reveal that the network structures of those glasses that are accessible by either preparation method are essentially identical. However, the significantly extended glass-forming ranges available by the sol-gel route facilitate exploration of the structure/composition relationships in more detail, revealing a number of interesting universal features throughout the whole glass system. Both short- and medium-range order appear to be controlled strongly by the O/P ratio of the glasses studied: Up to an O/P ratio of 3.5 (pyrophosphate composition), aluminum is predominantly six-coordinated and fully connected to phosphorus (Al(OP)6 sites). In the region 3.5 e O/P e 4.0, a dramatic structural transformation takes place, leading to the appearance of additional four- and fivecoordinated aluminum species whose second coordination spheres are also entirely dominated by phosphorus. The structure of glasses with an O/P ratio of precisely 4.0 (orthophosphate) is dominated by Al(OP)4 units. As the O/P ratio increases beyond 4.0, the average extent of Al-O-P connectivity is decreased significantly. Here, new types of five- and six-coordinated aluminum units, which are only weakly connected to ph osphorus, are formed, while the network modifier is attracted mainly by the phosphate units.
JMR, Bodenhausen, Quadrupolar transfer pathways
Quadrupolar transfer pathways
Sasa Antonijevic and Geoffrey Bodenhausen
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 297-304
Communication
aLaboratoire de Résonance Magnétique Biomoléculaire, Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne, Batochime, CH-1015 Lausanne, Switzerland
bDépartement de Chimie, associé au CNRS, Ecole Normale Supérieure, 24 rue Lhomond 75231, Paris Cedex 05, France
Abstract
A set of graphical conventions called quadrupolar transfer pathways is proposed to describe a wide range of experiments designed for the study of quadrupolar nuclei with spin quantum numbers I = 1, 3/2, 2, 5/2, etc. These pathways, which inter alea allow one to appreciate the distinction between quadrupolar and Zeeman echoes, represent a generalization of the well-known coherence transfer pathways. Quadrupolar transfer pathways not merely distinguish coherences with different orders -2I less-than-or-equals, slant p less-than-or-equals, slant +2I, but allow one to follow the fate of coherences associated with single transitions that have the same coherence order Click to view the MathML source but can be distinguished by a satellite order Click to view the MathML source.
Keywords: Coherence transfer pathways; Coherence order; Satellite order; Phase cycling; Quadrupolar echo
Sasa Antonijevic and Geoffrey Bodenhausen
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 297-304
Communication
aLaboratoire de Résonance Magnétique Biomoléculaire, Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne, Batochime, CH-1015 Lausanne, Switzerland
bDépartement de Chimie, associé au CNRS, Ecole Normale Supérieure, 24 rue Lhomond 75231, Paris Cedex 05, France
Abstract
A set of graphical conventions called quadrupolar transfer pathways is proposed to describe a wide range of experiments designed for the study of quadrupolar nuclei with spin quantum numbers I = 1, 3/2, 2, 5/2, etc. These pathways, which inter alea allow one to appreciate the distinction between quadrupolar and Zeeman echoes, represent a generalization of the well-known coherence transfer pathways. Quadrupolar transfer pathways not merely distinguish coherences with different orders -2I less-than-or-equals, slant p less-than-or-equals, slant +2I, but allow one to follow the fate of coherences associated with single transitions that have the same coherence order Click to view the MathML source but can be distinguished by a satellite order Click to view the MathML source.
Keywords: Coherence transfer pathways; Coherence order; Satellite order; Phase cycling; Quadrupolar echo
JMR, Amoreux, spin-9/2 signal enhancement
Resolution enhancement in 1D solid-state NMR spectra of spin-9/2 quadrupolar nuclei
Jean-Paul Amoureux and Julien Trébosc
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 311-316
Communication
Abstract
NMR is an insensitive spectroscopy, which often requires numerous accumulations, especially for 2D high-resolution methods (MQMAS and STMAS) for quadrupolar nuclei in solids. This may be a very important limitation for the case of insensitive nuclei, where a 1D spectrum with better resolution than the central-transition is then highly desirable. This problem has been addressed for the case of spin-5/2 nuclei by the Double-Quantum Filtered Satellite Transition Spectroscopy: DQF-SATRAS-ST1. We extend this concept to the spin-9/2 nuclei with the SATRAS-ST2 method. This method allows the observation of 1D spectra with a much better resolution than that observed in the isotropic projection of 2D MQ/ST1-MAS spectra. This enhanced resolution results from the much smaller homogeneous broadening that occurs on the SATRAS-ST2 method as compared to MQ/ST1-MAS spectra. The main interest in this method is for well-crystallized samples.
Keywords: Magic-angle spinning NMR; Quadrupolar nuclei; Satellite-transition MAS; High-resolution; Homogeneous broadening
Jean-Paul Amoureux and Julien Trébosc
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 311-316
Communication
Abstract
NMR is an insensitive spectroscopy, which often requires numerous accumulations, especially for 2D high-resolution methods (MQMAS and STMAS) for quadrupolar nuclei in solids. This may be a very important limitation for the case of insensitive nuclei, where a 1D spectrum with better resolution than the central-transition is then highly desirable. This problem has been addressed for the case of spin-5/2 nuclei by the Double-Quantum Filtered Satellite Transition Spectroscopy: DQF-SATRAS-ST1. We extend this concept to the spin-9/2 nuclei with the SATRAS-ST2 method. This method allows the observation of 1D spectra with a much better resolution than that observed in the isotropic projection of 2D MQ/ST1-MAS spectra. This enhanced resolution results from the much smaller homogeneous broadening that occurs on the SATRAS-ST2 method as compared to MQ/ST1-MAS spectra. The main interest in this method is for well-crystallized samples.
Keywords: Magic-angle spinning NMR; Quadrupolar nuclei; Satellite-transition MAS; High-resolution; Homogeneous broadening
JMR, Kuchel, 23Na and 133Cs NMR as probes of alignment in macromolecules
Apparatus for rapid adjustment of the degree of alignment of NMR samples in aqueous media: Verification with residual quadrupolar splittings in Na and Cs spectra
Philip W. Kuchel, Bogdan E. Chapman, Norbert Müller, William A. Bubb, David J. Philp and Allan M. Torres
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 256-265
aSchool of Molecular and Microbial Biosciences, University of Sydney, NSW 2006, Australia
bInstitut für Chemie, Johannes Kepler Universität A-4040 Linz, Austria
Abstract
NMR spectra of 23Na+ and 133Cs+ in gelatine in a silicone rubber tube that was stretched to various extents showed remarkably reproducible resonance multiplicity. The relative intensities of the components of the split peaks had ratios, 3:4:3, and 7:12:15:16:15:12:7, respectively, that conformed with those predicted using a Mathematica program. The silicone-rubber tube was sealed at its lower end by a small rubber stopper and placed inside a thick-walled glass tube. Gelatine was injected in solution into the silicone tube and ‘set’ by cooling below 30 °C. A plastic thumb-screw held the silicone tube at various degrees of extension, up to not, vert, similar2-fold. After constituting the gel in buffers containing NaCl and CsCl, both 23Na and 133Cs NMR spectroscopy revealed that after stretching the initial single Lorentzian line was split into a well-resolved triplet and a heptet, respectively. This was interpreted as being due to coupling between the electric quadrupoles of the nuclei and the average electric field gradient tensor of the collagen molecules of gelatine; these molecules became progressively more aligned in the direction of the main magnetic field, B0, of the vertical bore magnet, as the gel was stretched. This apparatus provides a simple way of demonstrating fundamental physical characteristics of quadrupolar cations, some characteristics of gelatine under stretching, and a way to invoke static distortion of red blood cells. It should be useful with these and other cell types, for studies of metabolic and membrane transport characteristics that may change when the cells are distorted, and possibly for structural studies of macromolecules.
Keywords: Alignment tensor; Electric field tensor; Quadrupolar splitting; Gelatine; Gel; Erythrocytes; 133-Caesium; 23-Sodium
Philip W. Kuchel, Bogdan E. Chapman, Norbert Müller, William A. Bubb, David J. Philp and Allan M. Torres
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 256-265
aSchool of Molecular and Microbial Biosciences, University of Sydney, NSW 2006, Australia
bInstitut für Chemie, Johannes Kepler Universität A-4040 Linz, Austria
Abstract
NMR spectra of 23Na+ and 133Cs+ in gelatine in a silicone rubber tube that was stretched to various extents showed remarkably reproducible resonance multiplicity. The relative intensities of the components of the split peaks had ratios, 3:4:3, and 7:12:15:16:15:12:7, respectively, that conformed with those predicted using a Mathematica program. The silicone-rubber tube was sealed at its lower end by a small rubber stopper and placed inside a thick-walled glass tube. Gelatine was injected in solution into the silicone tube and ‘set’ by cooling below 30 °C. A plastic thumb-screw held the silicone tube at various degrees of extension, up to not, vert, similar2-fold. After constituting the gel in buffers containing NaCl and CsCl, both 23Na and 133Cs NMR spectroscopy revealed that after stretching the initial single Lorentzian line was split into a well-resolved triplet and a heptet, respectively. This was interpreted as being due to coupling between the electric quadrupoles of the nuclei and the average electric field gradient tensor of the collagen molecules of gelatine; these molecules became progressively more aligned in the direction of the main magnetic field, B0, of the vertical bore magnet, as the gel was stretched. This apparatus provides a simple way of demonstrating fundamental physical characteristics of quadrupolar cations, some characteristics of gelatine under stretching, and a way to invoke static distortion of red blood cells. It should be useful with these and other cell types, for studies of metabolic and membrane transport characteristics that may change when the cells are distorted, and possibly for structural studies of macromolecules.
Keywords: Alignment tensor; Electric field tensor; Quadrupolar splitting; Gelatine; Gel; Erythrocytes; 133-Caesium; 23-Sodium
JMR, Christian Fernandez, XRD & 31P-31P, FSLG on microporous aluminophosphate IST-1
Characterization of microporous aluminophosphate IST-1 using H Lee–Goldburg techniques
Luís Mafra, João Rocha, Christian Fernandez and Filipe A. Almeida Paz
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 236-244
aDepartment of Chemistry, University of Aveiro, CICECO, 3810-193 Aveiro, Portugal
bLaboratoire Catalyse et Spectrochimie (CNRS UMR 6506), ENSICAEN and Université de Caen-Basse Normandie, 14050 Caen, France
Abstract
The presence of two independent methylamine species in microporous aluminophosphate IST-1 (|(CH3NH2)4(CH3NH+3)4(OH-)4|[Al12P12O48]) has been shown previously by synchrotron powder X-ray diffraction. One of these species, [N(1)–C(1)], links to a six-coordinated framework Al-atom [Al (1)], while the other methylamine [N(2)–C(2)] is protonated and hydrogen-bonded to three O-atoms [O (1), O (2) and O (12)]. We revisit the structure of IST-1 and report the complete assignment of the 1H NMR spectra by combining X-ray data and high-resolution heteronuclear/homonuclear solid-state NMR techniques based on frequency-switched Lee–Goldburg homonuclear decoupling and 31P–31P homonuclear recoupling. Careful analysis of the 2D 1H–X homonuclear correlation (X = 1H) and 2D heteronuclear correlation (X = 13C, 31P and 27Al) spectra allowed the distinction of both methylamine species and the assignment of all 31P and 13C resonances. For the first time at a relatively high (9.4 T) magnetic field, symmetric doublet patterns have been observed in the 13C spectra, caused by the influence of the 14N second-order quadrupolar interaction.
Keywords: FS-LG; 2D HETCOR MAS NMR; 31P DQ; Inorganic–organic hybrids; Templates; Microporous aluminophosphates; Methylamine
Luís Mafra, João Rocha, Christian Fernandez and Filipe A. Almeida Paz
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 236-244
aDepartment of Chemistry, University of Aveiro, CICECO, 3810-193 Aveiro, Portugal
bLaboratoire Catalyse et Spectrochimie (CNRS UMR 6506), ENSICAEN and Université de Caen-Basse Normandie, 14050 Caen, France
Abstract
The presence of two independent methylamine species in microporous aluminophosphate IST-1 (|(CH3NH2)4(CH3NH+3)4(OH-)4|[Al12P12O48]) has been shown previously by synchrotron powder X-ray diffraction. One of these species, [N(1)–C(1)], links to a six-coordinated framework Al-atom [Al (1)], while the other methylamine [N(2)–C(2)] is protonated and hydrogen-bonded to three O-atoms [O (1), O (2) and O (12)]. We revisit the structure of IST-1 and report the complete assignment of the 1H NMR spectra by combining X-ray data and high-resolution heteronuclear/homonuclear solid-state NMR techniques based on frequency-switched Lee–Goldburg homonuclear decoupling and 31P–31P homonuclear recoupling. Careful analysis of the 2D 1H–X homonuclear correlation (X = 1H) and 2D heteronuclear correlation (X = 13C, 31P and 27Al) spectra allowed the distinction of both methylamine species and the assignment of all 31P and 13C resonances. For the first time at a relatively high (9.4 T) magnetic field, symmetric doublet patterns have been observed in the 13C spectra, caused by the influence of the 14N second-order quadrupolar interaction.
Keywords: FS-LG; 2D HETCOR MAS NMR; 31P DQ; Inorganic–organic hybrids; Templates; Microporous aluminophosphates; Methylamine
JMR, Jakobsen, 33S SATRAS NMR, variable-temperature
Journal of Magnetic Resonance
Volume 180, Issue 2 , June 2006, Pages 170-177
Satellite transitions in natural abundance solid-state 33S MAS NMR of alums—Sign change with zero-crossing of CQ in a variable temperature study
Hans J. Jakobsen, Anders R. Hove, Henrik Bildsøe and Jørgen Skibsted
Instrument Center for Solid-State NMR Spectroscopy, Department of Chemistry, University of Aarhus, DK-8000 Aarhus C, Denmark
Abstract
Experiences obtained from recent improvements in the performance of solid-state 14N MAS NMR spectroscopy have been used in a natural abundance 33S MAS NMR investigation of the satellite transitions for this interesting spin I = 3/2 isotope. This study reports the first observation of manifolds of spinning sidebands for these transitions in 33S MAS NMR as observed for the two alums XAl(SO4)2·12H2O with X = NH4 and K. For the NH4-alum a variable temperature 33S MAS NMR study, employing the satellite transitions, shows that the 33S quadrupole coupling constant (CQ) exhibits a linear temperature dependence (in the range -35 °C to 70 °C) with a temperature gradient of 3.1 kHz/°C and undergoes a sign change with zero-crossing for CQ at 4 °C (277 K). For the isostructural K-alum a quite similar increase in the magnitude of CQ with increasing temperature is observed, and with a temperature gradient of 2.3 kHz/°C. Finally, for optimization purposes, a study on the effect of the applied pulse widths at constant rf field strength on the intensity and variation in second-order quadrupolar lineshape for the central (1/2 ? -1/2) transition of the K-alum has been performed.
Keywords: 33S MAS NMR; Satellite transitions; Alums; Variable-temperature NMR; Sign change of CQ; Simulations
Volume 180, Issue 2 , June 2006, Pages 170-177
Satellite transitions in natural abundance solid-state 33S MAS NMR of alums—Sign change with zero-crossing of CQ in a variable temperature study
Hans J. Jakobsen, Anders R. Hove, Henrik Bildsøe and Jørgen Skibsted
Instrument Center for Solid-State NMR Spectroscopy, Department of Chemistry, University of Aarhus, DK-8000 Aarhus C, Denmark
Abstract
Experiences obtained from recent improvements in the performance of solid-state 14N MAS NMR spectroscopy have been used in a natural abundance 33S MAS NMR investigation of the satellite transitions for this interesting spin I = 3/2 isotope. This study reports the first observation of manifolds of spinning sidebands for these transitions in 33S MAS NMR as observed for the two alums XAl(SO4)2·12H2O with X = NH4 and K. For the NH4-alum a variable temperature 33S MAS NMR study, employing the satellite transitions, shows that the 33S quadrupole coupling constant (CQ) exhibits a linear temperature dependence (in the range -35 °C to 70 °C) with a temperature gradient of 3.1 kHz/°C and undergoes a sign change with zero-crossing for CQ at 4 °C (277 K). For the isostructural K-alum a quite similar increase in the magnitude of CQ with increasing temperature is observed, and with a temperature gradient of 2.3 kHz/°C. Finally, for optimization purposes, a study on the effect of the applied pulse widths at constant rf field strength on the intensity and variation in second-order quadrupolar lineshape for the central (1/2 ? -1/2) transition of the K-alum has been performed.
Keywords: 33S MAS NMR; Satellite transitions; Alums; Variable-temperature NMR; Sign change of CQ; Simulations
PCCP: Wasylishen & Willans, 1J(X,35/37Cl) in triphenyl group-14 chlorides, experimental & ZORA DFT
Physical Chemistry Chemical Physics, 2006, (Advance Article)
DOI: 10.1039/b603937e
An NMR and relativistic DFT investigation of one-bond nuclear spin–spin coupling in solid triphenyl group-14 chlorides
Mathew J. Willans, Bryan A. Demko and Roderick E. Wasylishen
A solid-state nuclear magnetic resonance and zeroth-order regular approximation density functional theory, ZORA-DFT, study of one-bond nuclear spin–spin coupling between group-14 nuclei and quadrupolar 35/37Cl nuclei in triphenyl group-14 chlorides, Ph3XCl (X = C, Si, Ge, Sn and Pb), is presented. This represents the first combined experimental and theoretical systematic study of spin–spin coupling involving spin-pairs containing quadrupolar nuclei. Solid-state NMR spectra have been acquired for all compounds in which X has a spin-1/2 isotope—13C, 29Si, [117/119]Sn and 207Pb—at applied magnetic fields of 4.70, 7.05 and 11.75 T. From simulations of these spectra, values describing the indirect spin–spin coupling tensor—the isotropic indirect spin–spin coupling constant, 1J(X,35/37Cl)iso and the anisotropy of the J tensor, 1J(X,35/37Cl)—have been determined for all but the lead–chlorine spin-pair. To better compare the indirect spin–spin coupling parameters between spin-pairs, 1Jiso and 1J values were converted to their reduced coupling constants, 1Kiso and 1K. From experiment, the sign of 1Kiso was found to be negative while the sign of 1K is positive for all spin-pairs investigated. The magnitude of both 1Kiso and 1K was found to increase as one moves down group-14. Theoretical values of the magnitude and sign of 1Kiso and 1K were obtained from ZORA-DFT calculations and are in agreement with the available experimental data. From the calculations, the Fermi-contact mechanism was determined to provide the largest contribution to 1Kiso for all spin-pairs while spin-dipolar and paramagnetic spin–orbit mechanisms make significant contributions to the anisotropy of K. The inclusion of relativistic effects was found to influence K(Sn,Cl) and K(Pb,Cl).
DOI: 10.1039/b603937e
An NMR and relativistic DFT investigation of one-bond nuclear spin–spin coupling in solid triphenyl group-14 chlorides
Mathew J. Willans, Bryan A. Demko and Roderick E. Wasylishen
A solid-state nuclear magnetic resonance and zeroth-order regular approximation density functional theory, ZORA-DFT, study of one-bond nuclear spin–spin coupling between group-14 nuclei and quadrupolar 35/37Cl nuclei in triphenyl group-14 chlorides, Ph3XCl (X = C, Si, Ge, Sn and Pb), is presented. This represents the first combined experimental and theoretical systematic study of spin–spin coupling involving spin-pairs containing quadrupolar nuclei. Solid-state NMR spectra have been acquired for all compounds in which X has a spin-1/2 isotope—13C, 29Si, [117/119]Sn and 207Pb—at applied magnetic fields of 4.70, 7.05 and 11.75 T. From simulations of these spectra, values describing the indirect spin–spin coupling tensor—the isotropic indirect spin–spin coupling constant, 1J(X,35/37Cl)iso and the anisotropy of the J tensor, 1J(X,35/37Cl)—have been determined for all but the lead–chlorine spin-pair. To better compare the indirect spin–spin coupling parameters between spin-pairs, 1Jiso and 1J values were converted to their reduced coupling constants, 1Kiso and 1K. From experiment, the sign of 1Kiso was found to be negative while the sign of 1K is positive for all spin-pairs investigated. The magnitude of both 1Kiso and 1K was found to increase as one moves down group-14. Theoretical values of the magnitude and sign of 1Kiso and 1K were obtained from ZORA-DFT calculations and are in agreement with the available experimental data. From the calculations, the Fermi-contact mechanism was determined to provide the largest contribution to 1Kiso for all spin-pairs while spin-dipolar and paramagnetic spin–orbit mechanisms make significant contributions to the anisotropy of K. The inclusion of relativistic effects was found to influence K(Sn,Cl) and K(Pb,Cl).
JPC B: Acidity of Mesoporous MoOx/ZrO2 and WOx/ZrO2 Materials
J. Phys. Chem. B, ASAP Article 10.1021/jp0614087 S1520-6106(06)01408-8
Acidity of Mesoporous MoOx/ZrO2 and WOx/ZrO2 Materials: A Combined Solid-State NMR and Theoretical Calculation Study
Jun Xu, Anmin Zheng, Jun Yang, Yongchao Su, Jiqing Wang, Danlin Zeng, Mingjin Zhang,* Chaohui Ye, and Feng Deng*
State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Insitute of Physics and Mathematics, the Chinese Academy of Sciences, Wuhan 430071, P. R. China, and Department of Chemistry, Wuhan University of Science and Technology, Wuhan 430081, P. R. China
Abstract:
The acidity of mesoporous MoOx/ZrO2 and WOx/ZrO2 materials was studied in detail by multinuclear solid-state NMR techniques as well as DFT quantum chemical calculations. The 1H MAS NMR experiments clearly revealed the presence of two different types of strong Brnsted acid sites on both MoOx/ZrO2 and WOx/ZrO2 mesoporous materials, which were able to prontonate adsorbed pyrine-d5 (resulting in 1H NMR signals at chemical shifts in the range 16-19 ppm) as well as adsorbed trimethylphosphine (giving rise to 31P NMR signal at ca. 0 ppm). The 13C NMR of adsorbed 2-13C-acetone indicated that the average Brnsted acid strength of the two mesoporous materials was stronger than that of zeolite HZSM-5 but still weaker than that of 100% H2SO4, which was in good agreement with theoretical predictions. The quantum chemical calculations revealed the detailed structures of the two distinct types of Brnsted acid sites formed on the mesoporous MoOx/ZrO2 and WOx/ZrO2. The existence of both monomer and oligomer Mo (or W) species containing a Mo-OH-Zr (or W-OH-Zr) bridging OH group was confirmed with the former having an acid strength close to zeolite HZSM-5, with the latter having an acid strength similar to sulfated zirconia. On the basis of our NMR experimental and theoretical calculation results, a possible mechanism was proposed for the formation of acid sites on these mesoporous materials.
Acidity of Mesoporous MoOx/ZrO2 and WOx/ZrO2 Materials: A Combined Solid-State NMR and Theoretical Calculation Study
Jun Xu, Anmin Zheng, Jun Yang, Yongchao Su, Jiqing Wang, Danlin Zeng, Mingjin Zhang,* Chaohui Ye, and Feng Deng*
State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Insitute of Physics and Mathematics, the Chinese Academy of Sciences, Wuhan 430071, P. R. China, and Department of Chemistry, Wuhan University of Science and Technology, Wuhan 430081, P. R. China
Abstract:
The acidity of mesoporous MoOx/ZrO2 and WOx/ZrO2 materials was studied in detail by multinuclear solid-state NMR techniques as well as DFT quantum chemical calculations. The 1H MAS NMR experiments clearly revealed the presence of two different types of strong Brnsted acid sites on both MoOx/ZrO2 and WOx/ZrO2 mesoporous materials, which were able to prontonate adsorbed pyrine-d5 (resulting in 1H NMR signals at chemical shifts in the range 16-19 ppm) as well as adsorbed trimethylphosphine (giving rise to 31P NMR signal at ca. 0 ppm). The 13C NMR of adsorbed 2-13C-acetone indicated that the average Brnsted acid strength of the two mesoporous materials was stronger than that of zeolite HZSM-5 but still weaker than that of 100% H2SO4, which was in good agreement with theoretical predictions. The quantum chemical calculations revealed the detailed structures of the two distinct types of Brnsted acid sites formed on the mesoporous MoOx/ZrO2 and WOx/ZrO2. The existence of both monomer and oligomer Mo (or W) species containing a Mo-OH-Zr (or W-OH-Zr) bridging OH group was confirmed with the former having an acid strength close to zeolite HZSM-5, with the latter having an acid strength similar to sulfated zirconia. On the basis of our NMR experimental and theoretical calculation results, a possible mechanism was proposed for the formation of acid sites on these mesoporous materials.
Monday, May 15, 2006
Angewandte - Volume 45, Issue 18
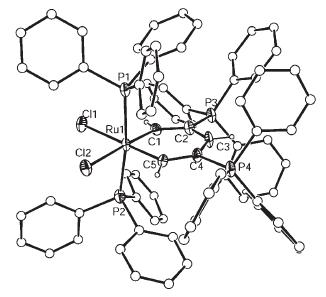
Wednesday, May 10, 2006
JACS - Volume 128, Issue 16
Cory's Comment: Using 2H SSNMR to determine the orientation of cholesterol in membranes as a function of chain unsaturation.
Title: Molecular Organization of Cholesterol in Unsaturated Phosphatidylethanolamines: X-ray Diffraction and Solid State 2H NMR Reveal Differences with Phosphatidylcholines
Authors: Saame Raza Shaikh, Vadim Cherezov, Martin Caffrey, Smita P. Soni, Daniel LoCascio, William Stillwell, and Stephen R. Wassall
Page #: 5375
Abstract: The major mammalian plasma membrane lipids are phosphatidylcholines (PCs), phosphatidylethanolamines (PEs), and cholesterol. Whereas PC-cholesterol interactions are well studied, far less is known about those between PE and cholesterol. Here, we investigated the molecular organization of cholesterol in PEs that vary in their degree of acyl chain unsaturation. For heteroacid sn-1 saturated (palmitoyl), sn-2 unsaturated (various acyl chain) PEs, cholesterol solubility determined by X-ray diffraction was essentially identical with 1 (oleoyl, 51 ± 3 mol %) and 2 (linoleoyl, 49 ± 2 mol %) double bonds before decreasing progressively with 4 (arachidonyl, 41 ± 3 mol %) and 6 (docosahexaenoyl, 31 ± 3 mol %) double bonds. With 6 double bonds in each chain, cholesterol solubility was further reduced to 8.5 ± 1 mol %. However, 2H NMR experiments established that the orientation of cholesterol in the same heteroacid PE membranes was unaffected by the degree of acyl chain unsaturation. A tilt angle of 15 ± 1 was measured when equimolar [3-2H1]cholesterol was added, regardless of the number of double bonds in the sn-2 chain. The finding that solubility of cholesterol in sn-1 saturated PEs depends on the amount of polyunsaturation in the sn-2 chain of PE differs from the equivalent PCs that universally incorporate ~50 mol % sterol. Unlike PCs, a differential in affinity for cholesterol and tendency to drive lateral segregation is inferred between polyunsaturated PEs. This distinction may have biological implications reflected by the health benefits of dietary polyunsaturated fatty acids that are often taken up into PE > PC.
MRC, Frydman, 23Na SSNMR of sodium sites in Na-nucleotide complexes
Magnetic Resonance in Chemistry
Vol: 44, Issue: 3, March 2006
pp. 366 - 374
Title: Solid-state NMR investigation of sodium nucleotide complexes
Authors: Grant, Christopher V.a; McElheny, Danb; Frydman, Veronicac; Frydman, Lucioc
Affiliations: a. Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093, USA
b. Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, IL 60637, USA
c. Weizmann Institute of Science, Rehovot, 76100, Israel
Keywords: NMR; MQMAS; 23Na spectra; ATP; nucleotide binding motifs; REDOR; quadrupolar relaxation
Abstract (English):
Solid-state NMR has been used to analyze the chemical environments of sodium sites in powdered crystalline samples of sodium nucleotide complexes. Three of the studied complexes have been previously characterized structurally by crystallography (disodium deoxycytidine-5’-monophosphate heptahydrate, disodium deoxyuridine-5’-monophosphate pentahydrate and disodium adensoine-5’-triphosphate trihydrate). For these salts, the nuclear quadrupole coupling parameters measured by 23Na multiple-quantum magic-angle-spinning NMR could be readily correlated with sodium ion coordination environments. Furthermore, two complexes that had not been previously characterized structurally, disodium uridine-3’-monophosphate and a disodium uridine-3’-monophosphate/disodium uridine-2’-monophosphate mix, were identified by solid-state NMR. A spectroscopic assignment of the four sites of an additional salt, disodium adensoine-5’-triphosphate trihydrate, is also presented and discussed within the context of creating a general approach for the spectroscopic assignment of multiple sites in sodium nucleotide complexes. Copyright © 2006 John Wiley & Sons, Ltd.
Vol: 44, Issue: 3, March 2006
pp. 366 - 374
Title: Solid-state NMR investigation of sodium nucleotide complexes
Authors: Grant, Christopher V.a; McElheny, Danb; Frydman, Veronicac; Frydman, Lucioc
Affiliations: a. Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093, USA
b. Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, IL 60637, USA
c. Weizmann Institute of Science, Rehovot, 76100, Israel
Keywords: NMR; MQMAS; 23Na spectra; ATP; nucleotide binding motifs; REDOR; quadrupolar relaxation
Abstract (English):
Solid-state NMR has been used to analyze the chemical environments of sodium sites in powdered crystalline samples of sodium nucleotide complexes. Three of the studied complexes have been previously characterized structurally by crystallography (disodium deoxycytidine-5’-monophosphate heptahydrate, disodium deoxyuridine-5’-monophosphate pentahydrate and disodium adensoine-5’-triphosphate trihydrate). For these salts, the nuclear quadrupole coupling parameters measured by 23Na multiple-quantum magic-angle-spinning NMR could be readily correlated with sodium ion coordination environments. Furthermore, two complexes that had not been previously characterized structurally, disodium uridine-3’-monophosphate and a disodium uridine-3’-monophosphate/disodium uridine-2’-monophosphate mix, were identified by solid-state NMR. A spectroscopic assignment of the four sites of an additional salt, disodium adensoine-5’-triphosphate trihydrate, is also presented and discussed within the context of creating a general approach for the spectroscopic assignment of multiple sites in sodium nucleotide complexes. Copyright © 2006 John Wiley & Sons, Ltd.
MRC, Dybowski, 207Pb NMR
Magnetic Resonance in Chemistry
Vol: 44, Issue: 3, March 2006
pp. 357 - 365
Title: Solid-state 207Pb NMR studies of lead-group 16 and mixed transition-metal/lead-group 16 element-containing materials
Authors: Van Bramer, S. E.a; Glatfelter, A.b; Bai, S.b; Dybowski, C.b; Neue, G.c; Perry, D. L.d
Affiliations: a. Department of Chemistry, Widener University, Chester, PA 19013 USA
b. Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716 USA
c. Physical Chemistry, University of Dortmund, D‐44221 Dortmund, Germany
d. Lawrence Berkeley National Laboratory, University of California, Berkeley, CA 94720 USA
Keywords: 207Pb NMR; solid-state NMR; lead selenide; lead selenate; calcium plumbate; strontium plumbite; barium plumbite; lead borate; lead zirconate; lead tungstate; lead meta-tantalate; lead niobate; lead molybdate; lead meta-vanadate; lead sulfite; lead sulfate; materials
Abstract (English):
207Pb solid-state NMR studies have been conducted on binary lead-group 16 and mixed transition-metal/lead group 16 materials, correlating the NMR chemical shifts of the materials with their structures. The experimental results show that the 207Pb chemical shifts are strongly influenced by the local electronic structure. Data are reported for lead selenide, lead selenate, calcium plumbate, strontium plumbite, barium plumbite, lead borate, lead zirconate, lead tungstate, lead meta-tantalate, lead niobate, lead molybdate, lead meta-vanadate, lead sulfite, and lead sulfate. Copyright © 2006 John Wiley & Sons, Ltd.
Vol: 44, Issue: 3, March 2006
pp. 357 - 365
Title: Solid-state 207Pb NMR studies of lead-group 16 and mixed transition-metal/lead-group 16 element-containing materials
Authors: Van Bramer, S. E.a; Glatfelter, A.b; Bai, S.b; Dybowski, C.b; Neue, G.c; Perry, D. L.d
Affiliations: a. Department of Chemistry, Widener University, Chester, PA 19013 USA
b. Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716 USA
c. Physical Chemistry, University of Dortmund, D‐44221 Dortmund, Germany
d. Lawrence Berkeley National Laboratory, University of California, Berkeley, CA 94720 USA
Keywords: 207Pb NMR; solid-state NMR; lead selenide; lead selenate; calcium plumbate; strontium plumbite; barium plumbite; lead borate; lead zirconate; lead tungstate; lead meta-tantalate; lead niobate; lead molybdate; lead meta-vanadate; lead sulfite; lead sulfate; materials
Abstract (English):
207Pb solid-state NMR studies have been conducted on binary lead-group 16 and mixed transition-metal/lead group 16 materials, correlating the NMR chemical shifts of the materials with their structures. The experimental results show that the 207Pb chemical shifts are strongly influenced by the local electronic structure. Data are reported for lead selenide, lead selenate, calcium plumbate, strontium plumbite, barium plumbite, lead borate, lead zirconate, lead tungstate, lead meta-tantalate, lead niobate, lead molybdate, lead meta-vanadate, lead sulfite, and lead sulfate. Copyright © 2006 John Wiley & Sons, Ltd.
MRC, Jakobsen, 14N MAS NMR, ammounium salts
Magnetic Resonance in Chemistry
Vol: 44, Issue: 3, March 2006
pp. 348 - 356
Title: Solid-state 14N MAS NMR of ammonium ions as a spy to structural insights for ammonium salts
Authors: Jakobsen, Hans J.a; Hove, Anders R.a; Hazell, Rita G.b; Bildsøe, Henrika; Skibsted, Jørgena
Affiliations: a. Instrument Centre for Solid‐State NMR Spectroscopy, Department of Chemistry, University of Aarhus, DK‐8000 Aarhus C, Denmark
b. Division of Inorganic Chemistry, Department of Chemistry, University of Aarhus, DK‐8000 Aarhus C, Denmark
Keywords: NMR; 14N; solid state; MAS; ammonium salts; quadrupole coupling; crystal structure; trimethyl benzylammonium chloride
Abstract (English):
The high resolution offered by magic-angle spinning (MAS), when compared to the static condition in solid-state NMR of powders, has been used to full advantage in a 14N MAS NMR study of some ammonium salts: CH3NH3Cl, (NH4)2(COO)2·H2O, (CH3)3(C6H5CH2)NCl, (CH3)3(C6H5)NI, [(n-C4H9)4N]2Mo2O7, (NH4)2HPO4, and NH4H2PO4. It is shown that the high-quality 14N MAS NMR spectra, which can be obtained for these salts, allow determination of the 14N quadrupole coupling parameters, i.e. CQ (the quadrupole coupling constant) and Q (the asymmetry parameter), with very high precision. In particular, it is shown that precise CQ, Q parameters can be determined for at least two different 14N sites in case the individual spinning-sideband (ssb) intensities arise from a single manifold of ssbs, i.e. the ssbs for the two sites cannot be resolved. This feature of 14N MAS NMR, which is the first demonstration for manifolds of ssb in MAS NMR without the potential information from a central transition, becomes especially useful at the slow spinning frequencies (nr = 1000–1500 Hz) applied to some of the ammonium salts studied here. The detection of the number of sites has been confirmed by the corresponding crystal structures determined from single-crystal X-ray diffraction (XRD), either in this work for the unknown structure of benzyl trimethylammonium chloride or from reports in the literature. The magnitudes of the 14N quadrupole coupling constants for the ammonium salts studied here are in the range from CQ 20 kHz to 1 MHz while the asymmetry parameters span the full range 0 £ Q £ 1. Clearly, the 14N quadrupole coupling parameters (CQ, Q) for ammonium ions appear highly sensitive toward crystal structure and therefore appreciably more informative for the characterization of ammonium salts in comparison to the isotropic 14N (or 15N) chemical shifts.
Vol: 44, Issue: 3, March 2006
pp. 348 - 356
Title: Solid-state 14N MAS NMR of ammonium ions as a spy to structural insights for ammonium salts
Authors: Jakobsen, Hans J.a; Hove, Anders R.a; Hazell, Rita G.b; Bildsøe, Henrika; Skibsted, Jørgena
Affiliations: a. Instrument Centre for Solid‐State NMR Spectroscopy, Department of Chemistry, University of Aarhus, DK‐8000 Aarhus C, Denmark
b. Division of Inorganic Chemistry, Department of Chemistry, University of Aarhus, DK‐8000 Aarhus C, Denmark
Keywords: NMR; 14N; solid state; MAS; ammonium salts; quadrupole coupling; crystal structure; trimethyl benzylammonium chloride
Abstract (English):
The high resolution offered by magic-angle spinning (MAS), when compared to the static condition in solid-state NMR of powders, has been used to full advantage in a 14N MAS NMR study of some ammonium salts: CH3NH3Cl, (NH4)2(COO)2·H2O, (CH3)3(C6H5CH2)NCl, (CH3)3(C6H5)NI, [(n-C4H9)4N]2Mo2O7, (NH4)2HPO4, and NH4H2PO4. It is shown that the high-quality 14N MAS NMR spectra, which can be obtained for these salts, allow determination of the 14N quadrupole coupling parameters, i.e. CQ (the quadrupole coupling constant) and Q (the asymmetry parameter), with very high precision. In particular, it is shown that precise CQ, Q parameters can be determined for at least two different 14N sites in case the individual spinning-sideband (ssb) intensities arise from a single manifold of ssbs, i.e. the ssbs for the two sites cannot be resolved. This feature of 14N MAS NMR, which is the first demonstration for manifolds of ssb in MAS NMR without the potential information from a central transition, becomes especially useful at the slow spinning frequencies (nr = 1000–1500 Hz) applied to some of the ammonium salts studied here. The detection of the number of sites has been confirmed by the corresponding crystal structures determined from single-crystal X-ray diffraction (XRD), either in this work for the unknown structure of benzyl trimethylammonium chloride or from reports in the literature. The magnitudes of the 14N quadrupole coupling constants for the ammonium salts studied here are in the range from CQ 20 kHz to 1 MHz while the asymmetry parameters span the full range 0 £ Q £ 1. Clearly, the 14N quadrupole coupling parameters (CQ, Q) for ammonium ions appear highly sensitive toward crystal structure and therefore appreciably more informative for the characterization of ammonium salts in comparison to the isotropic 14N (or 15N) chemical shifts.
MRC, Bryce, Solid-state NMR spectroscopy of the quadrupolar halogens: Cl, Br, I
MAGNETIC RESONANCE IN CHEMISTRY
Magn. Reson. Chem. 2006; 44: 409–450
Published online 19 January 2006 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/mrc.1741
Review
Solid-state NMR spectroscopy of the quadrupolar halogens: chlorine-35/37, bromine-79/81, and iodine-127
David L. Bryce* and Gregory D. Sward
Department of Chemistry, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada
A thorough review of 35/37Cl, 79/81Br, and 127I solid-state nuclear magnetic resonance (SSNMR) data is presented. Isotropic chemical shifts (CS), quadrupolar coupling constants, and other available information on the magnitude and orientation of the CS and electric field gradient (EFG) tensors for chlorine, bromine, and iodine in diverse chemical compounds is tabulated on the basis of over 200 references. Our coverage is through July 2005. Special emphasis is placed on the information available from the study of powdered diamagnetic solids in high magnetic fields. Our survey indicates a recent notable increase in the number of applications of solid-state quadrupolar halogen NMR, particularly 35Cl NMR, as high magnetic fields have become more widely available to solid-state NMR spectroscopists. We conclude with an assessment of possible future directions for research involving 35/37Cl, 79/81Br, and 127I solid-state NMR spectroscopy.
Magn. Reson. Chem. 2006; 44: 409–450
Published online 19 January 2006 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/mrc.1741
Review
Solid-state NMR spectroscopy of the quadrupolar halogens: chlorine-35/37, bromine-79/81, and iodine-127
David L. Bryce* and Gregory D. Sward
Department of Chemistry, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada
A thorough review of 35/37Cl, 79/81Br, and 127I solid-state nuclear magnetic resonance (SSNMR) data is presented. Isotropic chemical shifts (CS), quadrupolar coupling constants, and other available information on the magnitude and orientation of the CS and electric field gradient (EFG) tensors for chlorine, bromine, and iodine in diverse chemical compounds is tabulated on the basis of over 200 references. Our coverage is through July 2005. Special emphasis is placed on the information available from the study of powdered diamagnetic solids in high magnetic fields. Our survey indicates a recent notable increase in the number of applications of solid-state quadrupolar halogen NMR, particularly 35Cl NMR, as high magnetic fields have become more widely available to solid-state NMR spectroscopists. We conclude with an assessment of possible future directions for research involving 35/37Cl, 79/81Br, and 127I solid-state NMR spectroscopy.
CPL, nanoparticles, 59Co NMR
Chemical Physics Letters
Volume 422, Issues 4-6 , 10 May 2006, Pages 402-405
Cobalt nanoparticles with preferential hcp structure: A confirmation by X-ray diffraction and NMR
Vladimir V. Matveev, Dmitry A. Baranov, Gleb Yu. Yurkov, Nikita G. Akatiev, Ivan P. Dotsenko and Sergey P. Gubin
Abstract
In this Letter, nanostructured materials consisting of cobalt nanoparticles and different matrixes and formed by universal thermal destruction method are presented. Ultradispersed polytetrafluoroethylene (UPTFE) and polyethylene were used as matrixes for surface or volume stabilization of nanoparticles. 59Co NMR and X-ray diffraction methods were used to determine the structure of cobalt nanoparticles. The NMR data confirmed mainly the presence of hcp-like crystalline structure.
Volume 422, Issues 4-6 , 10 May 2006, Pages 402-405
Cobalt nanoparticles with preferential hcp structure: A confirmation by X-ray diffraction and NMR
Vladimir V. Matveev, Dmitry A. Baranov, Gleb Yu. Yurkov, Nikita G. Akatiev, Ivan P. Dotsenko and Sergey P. Gubin
Abstract
In this Letter, nanostructured materials consisting of cobalt nanoparticles and different matrixes and formed by universal thermal destruction method are presented. Ultradispersed polytetrafluoroethylene (UPTFE) and polyethylene were used as matrixes for surface or volume stabilization of nanoparticles. 59Co NMR and X-ray diffraction methods were used to determine the structure of cobalt nanoparticles. The NMR data confirmed mainly the presence of hcp-like crystalline structure.
JACS, Wimperis and Ashbrook; NOE enhancement of 11B SSNMR spectra
J. Am. Chem. Soc., ASAP Article 10.1021/ja0610939 S0002-7863(06)01093-6
Web Release Date: May 9, 2006
Nuclear Overhauser Effect (NOE) Enhancement of 11B NMR Spectra of Borane Adducts in the Solid State
Sharon E. Ashbrook, Nicholas G. Dowell, Ivan Prokes, and Stephen Wimperis*
School of Chemistry and EaStCHEM, University of St Andrews, St Andrews KY16 9ST, United Kingdom, Institute of Neurology, University College London, London WC1N 3BG, United Kingdom, Department of Biochemistry, University of Oxford, Oxford OX1 3QU, United Kingdom, and Department of Chemistry and WestCHEM, University of Glasgow, Glasgow G12 8QQ, United Kingdom
Abstract:
A strong 11B {1H} nuclear Overhauser effect (NOE) enhancement can be observed in solid-state 11B NMR spectra of borane adducts, yielding fractional enhancements, fI{S} = (I - I0)/I0, of the magic angle spinning (MAS) NMR signal of up to 155%. This is an interesting and unusual observation as 11B (spin I = 3/2) is a quadrupolar nucleus and the corresponding NOE is completely absent in solution. More generally, it shows that the NOE may have a wider role to play in solid-state NMR studies of dynamics than has been envisaged hitherto.
Web Release Date: May 9, 2006
Nuclear Overhauser Effect (NOE) Enhancement of 11B NMR Spectra of Borane Adducts in the Solid State
Sharon E. Ashbrook, Nicholas G. Dowell, Ivan Prokes, and Stephen Wimperis*
School of Chemistry and EaStCHEM, University of St Andrews, St Andrews KY16 9ST, United Kingdom, Institute of Neurology, University College London, London WC1N 3BG, United Kingdom, Department of Biochemistry, University of Oxford, Oxford OX1 3QU, United Kingdom, and Department of Chemistry and WestCHEM, University of Glasgow, Glasgow G12 8QQ, United Kingdom
Abstract:
A strong 11B {1H} nuclear Overhauser effect (NOE) enhancement can be observed in solid-state 11B NMR spectra of borane adducts, yielding fractional enhancements, fI{S} = (I - I0)/I0, of the magic angle spinning (MAS) NMR signal of up to 155%. This is an interesting and unusual observation as 11B (spin I = 3/2) is a quadrupolar nucleus and the corresponding NOE is completely absent in solution. More generally, it shows that the NOE may have a wider role to play in solid-state NMR studies of dynamics than has been envisaged hitherto.
JPC B, Ashbrook and Farnan, 89Y MAS NMR of Y2Ti2-xSnxO7 Pyrochlores
J. Phys. Chem. B, ASAP Article 10.1021/jp060844q S1520-6106(06)00844-3
89Y Magic-Angle Spinning NMR of Y2Ti2-xSnxO7 Pyrochlores
Sharon E. Ashbrook,* Karl R. Whittle, Gregory R. Lumpkin, and Ian Farnan
School of Chemistry and EaStCHEM, University of St. Andrews, North Haugh, St. Andrews KY16 9ST, U.K., and Department of Earth Sciences, University of Cambridge, Downing Street, Cambridge CB2 3EQ, U.K.
Abstract:
The yttrium local environment in the series of pyrochlores Y2Ti2-xSnxO7 was studied using 89Y NMR. Oxides with the pyrochlore structure exhibit a range of interesting physical and chemical properties, resulting in many technological applications, including the encapsulation of lanthanide- and actinide-bearing radioactive waste. The use of the nonradioactive Y3+ cation provides a sensitive probe for any changes in the local structure and ordering with solid solution composition, through 89Y (I = 1/2) NMR. We confirm that a single pyrochlore phase is formed over the entire compositional range, with Y found only on the eight-coordinated A site. A significant (~15 ppm) chemical shift is observed for each Sn substituted into the Y second neighbor coordination environment. The spectral signal intensities of the possible combinations of Sn/Ti neighbors match those predicted statistically assuming a random distribution of Sn4+/Ti4+ on the six-coordinated pyrochlore B site.
89Y Magic-Angle Spinning NMR of Y2Ti2-xSnxO7 Pyrochlores
Sharon E. Ashbrook,* Karl R. Whittle, Gregory R. Lumpkin, and Ian Farnan
School of Chemistry and EaStCHEM, University of St. Andrews, North Haugh, St. Andrews KY16 9ST, U.K., and Department of Earth Sciences, University of Cambridge, Downing Street, Cambridge CB2 3EQ, U.K.
Abstract:
The yttrium local environment in the series of pyrochlores Y2Ti2-xSnxO7 was studied using 89Y NMR. Oxides with the pyrochlore structure exhibit a range of interesting physical and chemical properties, resulting in many technological applications, including the encapsulation of lanthanide- and actinide-bearing radioactive waste. The use of the nonradioactive Y3+ cation provides a sensitive probe for any changes in the local structure and ordering with solid solution composition, through 89Y (I = 1/2) NMR. We confirm that a single pyrochlore phase is formed over the entire compositional range, with Y found only on the eight-coordinated A site. A significant (~15 ppm) chemical shift is observed for each Sn substituted into the Y second neighbor coordination environment. The spectral signal intensities of the possible combinations of Sn/Ti neighbors match those predicted statistically assuming a random distribution of Sn4+/Ti4+ on the six-coordinated pyrochlore B site.
Tuesday, May 09, 2006
Chem Soc Rev, Sharon Ashbrook and Mark E. Smith, 17O review
Chemical Society Reviews, 2006, (Advance Article)
DOI: 10.1039/b514051j
Solid state 17O NMR—an introduction to the background principles and applications to inorganic materials
Sharon E. Ashbrook and Mark E. Smith
Oxygen is a key chemical element and solid state NMR can provide unique insight into the its local environment. In the last decade there have been significant advances (sensitivity, resolution) in the NMR methodology for non-integer spin quadrupole nuclei such as oxygen and the background to these techniques is presented in this tutorial review. The information that the NMR parameters can provide about the local environment is explained through a series of illustrations from different areas of solid state chemistry and structural science of inorganic materials.
DOI: 10.1039/b514051j
Solid state 17O NMR—an introduction to the background principles and applications to inorganic materials
Sharon E. Ashbrook and Mark E. Smith
Oxygen is a key chemical element and solid state NMR can provide unique insight into the its local environment. In the last decade there have been significant advances (sensitivity, resolution) in the NMR methodology for non-integer spin quadrupole nuclei such as oxygen and the background to these techniques is presented in this tutorial review. The information that the NMR parameters can provide about the local environment is explained through a series of illustrations from different areas of solid state chemistry and structural science of inorganic materials.
PCCP: Eden, CP for selective Cq's
Physical Chemistry Chemical Physics, 2006, 8, 1994 - 1999
DOI: 10.1039/b600757k
Quadrupolar coupling selective cross-polarization in solid state NMR
Mattias Edén
A new strategy is presented for achieving selective heteronuclear polarization transfers from half-integer quadrupolar spins in magic-angle spinning (MAS) NMR. By combining cross-polarization with a recently introduced RAPT pulse sequence that selectively excites the signal of a half-integer quadrupolar nucleus based on its quadrupolar coupling constant magnitude, we demonstrate that hetero-nuclei in its close proximity may be selectively excited. Selective 23Na 1H polarization transfers are demonstrated in Na2MoO4·2H2O, Na2HPO4·2H2O and a mixture of NaHCO3 and Na2HPO4·2H2O.
DOI: 10.1039/b600757k
Quadrupolar coupling selective cross-polarization in solid state NMR
Mattias Edén
A new strategy is presented for achieving selective heteronuclear polarization transfers from half-integer quadrupolar spins in magic-angle spinning (MAS) NMR. By combining cross-polarization with a recently introduced RAPT pulse sequence that selectively excites the signal of a half-integer quadrupolar nucleus based on its quadrupolar coupling constant magnitude, we demonstrate that hetero-nuclei in its close proximity may be selectively excited. Selective 23Na 1H polarization transfers are demonstrated in Na2MoO4·2H2O, Na2HPO4·2H2O and a mixture of NaHCO3 and Na2HPO4·2H2O.
Subscribe to:
Comments (Atom)

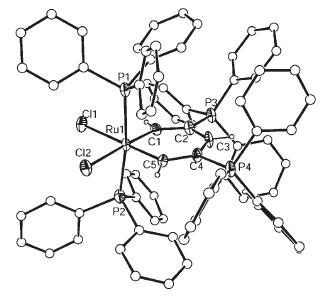{kind=link}